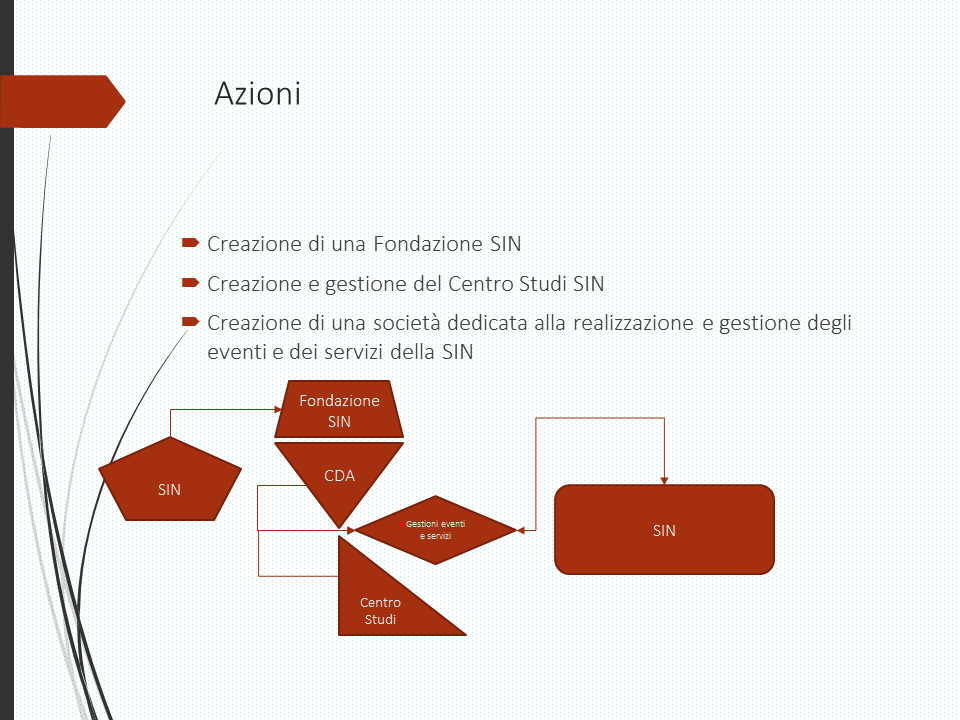
• SIN
La SIN è stata fondata nel 1907 ed ha lo scopo istituzionale di promuovere in Italia gli studi neurologici, finalizzati allo sviluppo della ricerca scientifica, alla formazione, all’aggiornamento degli specialisti e al miglioramento della qualità professionale nell’assistenza ai soggetti con malattie del sistema nervoso. La SIN è la società degli specialisti neurologi, sia che lavorino nel pubblico o nel privato, sul territorio, negli Ospedali o nell’Università. Il numero dei soci è notevolmente aumentato negli ultimi anni, superando nel 2008 il numero di 3000. Vogliamo ricordarti le principali iniziative SIN. Il Congresso annuale, che rappresenta oramai l’appuntamento annuale più importante di confronto scientifico e di aggiornamento professionale per i neurologi italiani e per la cui iscrizione i soci SIN hanno importanti agevolazioni, è diviso in diverse parti, alcune delle quali destinate alla libera comunicazione dei risultati conseguiti dai gruppi di ricerca, altre dedicate all’aggiornamento su specifici temi, decisi di anno in anno dal Consiglio Direttivo. La frequenza al Congresso da diritto ai crediti della Educazione Medica Continua, che vengono divisi nei singoli eventi Congressuali, in maniera tale che ognuno liberamente scelga quali sessioni frequentare. Particolare attenzione è da sempre dedicata ai giovani neurologi, che hanno a disposizione numerose borse per frequentare gratuitamente il Congresso. I gruppi di Studio e Associazioni autonome aderenti alla SIN, che sono oramai giunti al ragguardevole numero di 29, hanno spazi riservati al Congresso, con lo scopo di informare i neurologi sugli avanzamenti negli specifici settori dei quali si occupano.
La SIN ha una rivista ufficiale, Neurological Sciences, che ha raggiunto nel 2002 un Impact Factor molto vicino all’1 e che la colloca fra le riviste più importanti d’Europa. Iscrivendoti alla SIN, riceverai il Neurological Sciences gratuitamente a casa.
Il Sito SIN è organizzato in maniera tale da facilitare la navigazione del visitatore, che può accedere da ogni pagina del sito ad ogni altro argomento. Potrà così conoscere la storia della Società, il suo statuto, la composizione degli organi direttivi, le attività dei numerosi gruppi di studio e delle Associazioni autonome aderenti, i programmi delle Sezioni Regionali, i meeting e i Congressi patrocinati dalla Società che si terranno nei prossimi mesi, le linee guida promosse dalla Società, ed anche le offerte di lavoro nel mondo neurologico. Tutto questo per facilitare i contatti fra il visitatore e la SIN, a cui potrà fare domanda di iscrizione compilando l’apposito modulo scaricabile on line. Gli iscritti SIN potranno accedere alla parte riservata del sito mediante user e password.
• FONDAZIONE
Cos’è una Fondazione
- In base alla definizione coniata dall’European Foundation Centre di Bruxelles, una fondazione è un ente privato senza finalità di lucro con una propria sorgente di reddito che deriva normalmente – anzi, in Italia, necessariamente – da un Patrimonio.
- Questo ente, dotato di una propria organizzazione e di propri organi di governo, usa le proprie risorse finanziarie per scopi educativi, culturali, religiosi, sociali o altri scopi di pubblica utilità, sia sostenendo persone ed enti (fondazione di erogazione), sia organizzando e gestendo direttamente i suoi programmi (fondazione operativa).
- Una fondazione è costituita da un fondatore – anche più persone congiuntamente ovvero una persona giuridica – tramite un atto pubblico; la costituzione dell’ente deve essere sancita da un notaio tramite l’atto di fondazione, mentre per poter operare necessita di un riconoscimento giuridico che sottopone tutti gli atti della fondazione al controllo di legittimità di un’apposita autorità vigilante (art. 12 e seguenti del Codice Civile).
- Le principali norme organizzative per il corretto funzionamento dell’ente sono raccolte nello Statuto, che costituisce parte integrante dell’atto di fondazione.
- Il patrimonio è un elemento necessario, in quanto la legge e la giurisprudenza non ammettono fondazioni finanziate esclusivamente da contributi di terzi; la costituzione di una fondazione, pertanto, può essere vista come una immobilizzazione di risorse economiche e di conseguenza il legislatore si è preoccupato di garantire che tali risorse vengano utilizzate efficacemente ed efficientemente a beneficio della collettività.
- E’ per queste ragioni che l’autorità preposta al riconoscimento giuridico della fondazione è legittimata a richiedere un patrimonio minimo, tale da consentire l’effettiva possibilità di raggiungere lo scopo (congruità del patrimonio rispetto allo scopo).
- Per il riconoscimento nazionale il Ministero dell’Interno ha imposto una soglia minima di 100.000,00 euro, al di sotto della quale l’istanza di riconoscimento non viene accolta dalla prefettura competente a riceverla, è ammesso un capitale inferiore ai 100.000,00 euro solo se la fondazione dispone di un patrimonio immobiliare di una certa entità, valutata dal Ministero caso per caso; il Consiglio di Stato, comunque, nel corso della procedura di riconoscimento, può richiedere una soglia più elevata per fondazioni che operino in particolari settori o che comunque adottino una struttura operativa anziché di semplice erogazione.
Struttura organizzativa
Soci Fondatori
La Fondazione sarà promossa dai Fondatori che interverranno all’atto della costituzione e da soggetti che aderiranno e contribuiranno al patrimonio della Fondazione successivamente all’atto costitutivo.
Soci Sostenitori – Enti e Istituzioni
Alla fondazione possono aderire Università ed altri enti, in qualità di Sostenitori.
Le università contribuiscono con contributi scientifico/culturale, rendendo disponibili le proprie competenze. La fondazione potrà avere ulteriori Sostenitori, previa accettazione della relativa domanda da parte del Consiglio di Amministrazione.
Soci Ordinari – Aziende e Privati
Possono divenire soci ordinari le persone fisiche e giuridiche pubbliche o private e gli enti e organismi che ne facciano richiesta, condividendo gli scopi della Fondazione ed impegnandosi a contribuire al suo finanziamento, previa accettazione alla relativa domanda da parte del Consiglio di Amministrazione.
Organi della Fondazione
Gli organi della Fondazione saranno:
- Il Presidente
- Il Consiglio di Amministrazione
- Il Segretario Generale
- Il Comitato Tecnico Scientifico
- Il Collegio dei Revisori dei Conti
Per le funzioni svolte a qualsiasi titolo dal Presidente e dai componenti del Consiglio Generale e del Consiglio di Amministrazione non sono corrisposti emolumenti, fatto salvo il rimborso spese.
Principali adempimenti
- Redigere l’atto di fondazione, cioè l’atto costitutivo e lo statuto, secondo le norme previste dal Codice Civile e dalla legislazione inerente.
- Rivolgersi ad un notaio, perchè l’atto di fondazione assuma la veste di atto pubblico notarile, indispensabile per fondare una fondazione.
- Chiedere il riconoscimento della fondazione presso la prefettura di competenza, per fondazioni che operano a livello nazionale, o presso la regione, per le fondazioni che operano a livello regionale.
La fondazione può essere definita come una organizzazione costituita per la destinazione di un patrimonio privato, autonomo e vincolato, ad uno scopo di pubblica utilità. Attraverso tale ente i privati perseguono fini di interesse comune.
Se nell’associazione prevale l’elemento personale, cioè il vincolo tra un gruppo di persone che con le loro decisioni gestiscono l’ente e mirano al conseguimento di uno scopo, nelle fondazioni l’elemento centrale è il patrimonio, messo a disposizione dal fondatore, destinato e vincolato al conseguimento di uno scopo preciso.
Esistono due tipologie di fondazioni: quelle operative, che gestiscono in proprio una attività, come casa di cura, biblioteche, scuole, e le fondazioni di erogazione, che raggiungono il loro scopo limitandosi ad erogare sussidi e contributi ad enti o soggetti terzi.
A differenza delle associazioni, in cui l’atto costitutivo è un contratto tra più persone, l’atto costitutivo delle fondazioni è un’ atto unilaterale che può essere fatto anche da una sola persona o persona giuridica. L’atto dovrà avere specifici requisiti richiesti dalla legge (in merito leggi l’articolo sull’atto di fondazione).
Per costituire una fondazione, è necessario chiedere il riconoscimento all’autorità competente. Il riconoscimento ha efficacia costitutiva dell’ente e la sua principale conseguenza è l’acquisto della personalità giuridica (in merito leggi l’articolo sul riconoscimento della fondazione)
Nelle fondazioni, il fondatore, ovvero chi ha destinato i suoi beni ad uno specifico scopo, non concorrerà alla gestione dell’ente, che sarà invece gestito dagli amministratori, che avranno il compito di applicare ed eseguire l’atto di fondazione (anche se il fondatore può comunque riservarsi il diritto di partecipare alla gestione e all’amministrazione dell’ente).
Possono essere fondatori persone fisiche, persone giuridiche (società e imprese), enti collettivi ed enti pubblici.
Nello statuto deve esser specificato lo scopo della fondazione, mentre non è necessaria l’indicazione specifica delle attività poste in essere per raggiungere questo scopo.
Generalmente, gli amministratori possono scegliere liberamente il tipo di attività d a esercitare. comprese attività di natura economica e imprenditoriale. Comunque tali attività devono essere sempre finalizzate al perseguimento dello scopo. (in merito leggi l’articolo sugli amministratori della fondazione). In ogni caso, il fondatore può comunque delimitare l’attività della fondazione.
Per costituire una fondazione, il patrimonio dovrà essere adeguato al perseguimento dello scopo. Per tale motivo sono particolarmente diffuse le fondazioni costituite da imprese, società, o gruppi di persone, che dispongono di un certo capitale da conferire.
Atto di Fondazione
La fondazione nasce con l’atto di fondazione, ovvero un atto unilaterale di autonomia privata.
L’atto si distingue in un atto costitutivo, in cui viene manifestata la volontà del fondatore di costituire l’ente, e in uno statuto in cui si determinano le modalità di raggiungimento dello scopo e e l’organizzazione dell’ente.
L’atto di fondazione può essere redatto tra vivi oppure per testamento.
L’atto di fondazione deve avere la forma di atto pubblico notarile. L’atto contiene in sè una disposizione patrimoniale (conferimento del patrimonio all’ente) e l’istituzione di una specifica organizzazione per gestire lo stesso patrimonio. Il notaio deve poi, entro trenta giorni, denunciare al prefetto la formazione dell’atto di donazione.
L’atto di fondazione deve contenere:
- la denominazione dell’ente e lo scopo della fondazione, che deve essere determinato e lecito, e non in contrasto con norme imperative o con l’ordine pubblico. Inoltre deve coincidere con un interesse collettivo ed altruistico meritevole di tutela, e non con il mero interesse individuale del fondatore. Lo scopo dell’associazione può essere perseguito anche con attività economica ed imprenditoriale.
- la descrizione del patrimonio, che costituisce lo strumento essenziale per il conseguimento dello scopo della fondazione, oltre a servire come garanzia per gli eventuali creditori dell’ente. Il patrimonio deve essere adeguato allo scopo perseguito dall’ente e utilizzato solo per realizzare tale scopo.
- l’organizzazione di uomini e mezzi, che svolgono l’attività necessaria per il funzionamento dell’ente. L’organizzazione della fondazione è una struttura stabile di personale, organi e uffici, strumentali al perseguimento dello scopo dell’ente.
- le norme sull’ordinamento e l’amministrazione, cioè le regole che disciplinano la nomina e il funzionamento dell’organo amministrativo. Questo può essere composto da una o più persone, così da formare un consiglio di amministrazione. Tale ruolo può essere affidato anche a persone giuridiche 8 in merito leggi l’articolo sugli amministratori)
- l’atto di fondazione può prevedere anche altri organi collegiali, quali assemblee, consigli direttivi e organi di controllo. In tal caso l’atto di fondazione dovrà indicare le rispettive attribuzioni, nonchè le modalità di nomina e di sostituzione.
- i criteri e le modalità di erogazioni delle rendite a terzi, che devono essere determinate nell’atto di fondazione. Questo riguarda sole le fondazioni la cui attività consiste nell’erogare somme di denaro o altre prestazioni a favore dei beneficiari. Con il termine di rendita si intende sia il patrimonio originale della fondazione, sia i proventi della sua attività
- le norme relative all’estinzione dell’ente, alla sua trasformazione e alla devoluzione del patrimonio. In nessun caso, l’atto di fondazione può prevedere che i beni residui, in caso di estinzione dell’ente, tornino al fondatore o vadano ai suoi eredi.
Le Agevolazioni
Tutte le agevolazioni sono indicate nel Dlgs 460/1997 – pdf.
Ai fini delle imposte dirette, per esempio:
- non è considerata attività commerciale lo svolgimento di attività istituzionali
- non concorrono alla formazione della base imponibile i proventi derivanti dall’esercizio di attività connesse o le somme versate dagli associati o partecipanti a titolo di contributo o quote associative
- non concorrono alla formazione del reddito i fondi raccolti durante manifestazioni pubbliche occasionali, anche se in contropartita di beni di modico valore o di servizi
- non concorrono alla formazione del reddito i contributi corrisposti da amministrazioni pubbliche in regime convenzionale.
Per quanto riguarda l’Iva, per esempio, non c’è l’obbligo di ricevuta o scontrino fiscale per le operazioni riconducibili alle attività istituzionali.
Tra le agevolazioni riguardanti le altre imposte indirette rientrano:
- l’esenzione dall’imposta di bollo e della tassa sulle concessioni governative
- il pagamento dell’imposta di registro in misura fissa sugli atti traslativi a titolo oneroso della proprietà di beni immobili e degli atti traslativi o costitutivi di diritti reali immobiliari di godimento.
Esempi delle finalità di una Fondazione
- Promuovere la ricerca di base e clinica nel campo della neurologia
- Organizzare campagne di raccolta di fondi
- Affiancare e collaborare con altri Enti od Istituzioni che condividano la finalità di promozione della ricerca
- Organizzare eventi a scopo di informazione e divulgazione scientifica
- Organizzare eventi scientifici
- Eseguire consulenze professionali, svolgere attività di assistenza tecnica, supporto operativo e studi di fattibilità in ambito clinico, economico, organizzativo ed istituzionale – anche per conto di pubbliche amministrazioni, di strutture private e di organismi internazionali
- Promuovere ed organizzare campagne di stampa, televisive ed informatiche
- Promuovere ogni forma di iniziativa rivolta ad allargare conoscenze e consenso nel campo della ricerca sui disturbi neurologici
- Assegnazione di finanziamenti a progetti di ricerca
• CENTRO SUTDI
Organigramma
Incarichi Centro Studi SIN
Biennio 2023-2025
Presidente e Consiglio di amministrazione:
- Past President SIN Alfredo Berardelli
- Presidente SIN Alessandro Padovani
- Presidente Eletto SIN Mario Zappia
- Vicepresidente SIN Vincenzo Andreone
- Segretario SIN Nicola De Stefano
- Tesoriere SIN Claudio Gasperini
Segreteria Organizzativa
Barbara Frati
Il Coordinatore del Centro Studi, nominato dal Consiglio Direttivo, il 12 maggio 2025, è la Prof.ssa Antonella Conte, che affiancherà il Presidente nell’individuazione e gestione delle seguenti figure di riferimento per le diverse aree operative e obiettivi:
- Linee Guida e Biblioteca: Dott. Nicola De Stefano
- Rete dei Centri Collaborativi ed Epidemiologia: Prof. Paolo Ragonese
- Censimento Attività di Ricerca: Prof.ssa Antonella Conte
- Censimento Strutture Neurologiche: Dott. Rocco Quatrale
• SCUOLA
OBIETTIVI DELLA SCUOLA:
La Scuola Superiore di Neurologia SIN si pone come target la formazione di specializzandi e giovani neurologi prevedendo però anche uno spazio per l’aggiornamento del neurologo già professionalmente impegnato, sia universitario che ospedaliero.
Della Scuola Superiore di Neurologia dovrebbero poter fruire, per attività formative e di aggiornamento, le Associazioni Autonome aderenti alla SIN, i vari gruppi di Studio e tutte le articolazioni organizzative della SIN.
Per poter raggiungere questi obiettivi si potranno utilizzare quindi sia corsi residenziali che piattaforme digitali.
STRUTTURA DELLA SCUOLA:
La scuola Superiore di Neurologia è costituita da un Comitato Scientifico composto da: Prof. A. Padovani, Prof. A. Berardelli, Prof. M. Zappia, Prof. N. De Stefano, Dott. C. Gasperini.
La struttura operativa è costituita dalla segreteria SIN SienaCongress che si occuperà della parte logistico-organizzativa nonché della coordinazione fra tutte le figure di riferimento interessate (membri della commissione, partner Accademia della Medicina)
ORGANI DI GOVERNO
- Direttore Scientifico: ha la responsabilità della gestione della scuola, resta in carica 2 anni, ed è designato dal Consiglio Direttivo della SIN. Si propone che tale ruolo sia ricoperto dal Past President SIN, per garantire sempre una continuità di garanzia. Attualmente il Past President SIN è il Prof. Alfredo Berardelli.
Tuttavia, questa decisione dovrà essere presa in via definitiva alla luce dei risultati del primo bienno di attività della scuola. - Comitato Scientifico: è composto da 6/8 membri indicati dalla SIN che riflettono le diverse aree scientifiche in cui si articolano le neuroscienze. Nella sua prima applicazione il Comitato Scientifico è costituto dalla Commissione che ha curato la fondazione della Scuola. I membri del Comitato Scientifico cooperano alla realizzazione degli eventi interagendo con il l Presidente e con la Segreteria organizzativa in relazione alla proprie specifiche competenze d’area.
- Segretario Scientifico: questo ruolo rimane per il momento vacante.
• CONSIGLIO DIRETTIVO
Presidente: MARIO ZAPPIA
Direttore Dipartimento ad Attività Integrata di Scienze Neurologiche e della Ricerca Neurobiologica
Direttore U.O.C. Clinica Neurologica – A.O.U. “Policlinico-Vittorio Emanuele”
E-mail m.zappia@unict.it
Vice Presidente: CLAUDIO GASPERINI
Direttore UOC della Divisione di
Neurologia presso il Dipartimento di Neuroscienze Testa Collo dell’Ospedale S.Camillo-Forlanini di Roma, piazza Carlo Forlanini, 1 Roma
E-mail: c.gasperini@libero.it
Segretario: CRISTINA TASSORELLI
Professore Ordinario di Neurologia, Università di Pavia, Dipartimento di Scienze del Sistema Nervoso e del Comportamento Fondazione C. Mondino
Email: cristina.tassorelli@mondino.it
Presidente Eletto: NICOLA DE STEFANO
Università di Siena – Dipartimento di Medicina, Chirurgia e Neuroscienze – Viale Bracci 2, 53100 Siena; Tel. 0577235807; Fax 0577233411 E-mail destefano@unisi.it
Past President: ALESSANDRO PADOVANI
Università degli Studi di Brescia – Dip. di Scienze Cliniche Sperimentali
Clinica Neurologica – UO Neurologia 2 – ASST Spedali Civili – Policlinico Satellite Brescia
Tel. 0303995632, E-mail presidente@neuro.it
Consigliere: MARIA CONCETTA ALTAVISTA
Direttore Unità Operativa Complessa di Neurologia – ASL Roma 1
E-mail mc.altavista@gmail.com
Consigliere: PAOLO CALABRESI
Professore Ordinario di Neurologia e Direttore della Clinica Neurologica della Fondazione Policlinico Universitario Agostino Gemelli IRCCS Roma dell’Università Cattolica del Sacro Cuore, Largo Agostino Gemelli, 8 – 00168 Roma
E-mail: paolo.calabresi@policlinicogemelli.it
Consigliere: MASSIMO FILIPPI
Professore Ordinario di Neurologia presso l’Università Vita-Salute San Raffaele; Direttore della Scuola di Specializzazione in Neurologia e Presidente del Corso di Laurea in Fisioterapia della stessa Università. Direttore delle Unità Operative di Neurologia e Neuroriabilitazione, del Centro Sclerosi Multipla, del Servizio di Neurofisiologia e dell’Unità di Neuroimaging Quantitativo dell’Ospedale San Raffaele di Milano, Via Olgettina, 58, 20132 Milano.
E-mail: filippi.massimo@hsr.it
Consigliere: ANGELO LABATE
Professore Ordinario di Neurologia – Dipartimento Biomedicina, Neuroscienze e Diagnostica avanzata – Università degli Studi di Palermo
E-mail: angelo.labate@unipa.it
Consigliere: LETIZIA LEOCANI
Professoressa Associata di Neurologia – Università Vita-Salute San Raffaele di Milano
E-mail: leocani.letizia@unisr.it
Consigliere: MATILDE LEONARDI
Direttore della Struttura Complessa Neurologia, Salute Pubblica, Disabilità e del Coma Research Centre della Fondazione IRCCS Istituto Neurologico Carlo Besta di Milano, Via Celoria, 11 – 20133 Milano
E-mail: matilde.leonardi@istituto-besta.it
Consigliere: ROCCO LIGUORI
Professore Ordinario di Neurologia presso il Dipartimento di Scienze Biomediche e NeuroMotorie dell’Università di Bologna e Direttore dell’UOC Clinica Neurologica, IRCCS Istituto delle Scienze Neurologiche, Azienda USL di Bologna, Via Altura 3 – 40139 Bologna
Tel. 0514966917 – E-mail: rocco.liguori@unibo.it
Consigliere: LEONARDO LOPIANO
Professore Ordinario di Neurologia – Dipartimento di Neuroscienze dell’Università di Torino e Direttore della Divisione Universitaria di Neurologia 2 dell’A.O.U. Città della Salute e della Scienza di Torino.
E-mail: leonardo.lopiano@unito.it
Consigliere: ALESSANDRA NICOLETTI
Professore Ordinario di Neurologia presso l’Università di Catania, via Santa Sofia 78 – 95123 Catania
E-mail: anicolet@unict.it
Consigliere: MARIA TERESA PELLECCHIA
Professoressa Associata di Neurologia – Università degli Studi di Salerno e Responsabile del Programma UOC di “Neurologia Sperimentale” presso AOU San Giovanni di Dio e Ruggi d’Aragona di Salerno.
E-mail: mpellecchia@unisa.it
Consigliere: ANGELO QUARTATONE
Professore Ordinario di Neurologia – Università degli Studi di Messina.
E-mail: angelo.quartarone@irccsme.it
Consigliere: ANTONIO RUSSO
Professore Ordinario di Neurologia – Università degli Studi di Napoli “Luigi Vanvitelli” e Direttore del Programma Interdipartimentale di Medicina delle Cefalee e delle Sindromi Dolorose Primarie dell’AOU “Luigi Vanvitelli” di Napoli.
E-mail: dottor.russo@gmail.com
Consigliere: MARIA SESSA
Direttore della SC Neurologia & Stroke Unit dell’AAST Grande Ospedale Metropolitano Niguarda di Milano – Piazza dell’Ospedale Maggiore 3, 20162 Milano.
E-mail: maria.sessa@ospedaleniguarda.it
Consigliere: MICHELE TINAZZI
Professore Ordinario di Neurologia, Università di Verona e Direttore UOC Neurologia B, Azienda Ospedaliera-Universitaria Integrata (AOUI) di Verona.
E-mail: michele.tinazzi@univr.it
Consigliere: GINO VOLPI
Direttore della S.O.C. di Neurologia e Stroke Unit Neurologia di Pistoia.
E-mail: gino.volpi@uslcentro.toscana.it
Proboviro: ALFREDO BERARDELLI
Professore Ordinario di Neurologia, Dipartimento di Neuroscienze Umane, Policlinico Umberto I, Sapienza Università di Roma, Viale Università 30, Roma tel. 06/49914700; fax 06/49914074 alfredo.berardelli@uniroma1.it
Proboviro: GABRIELE SICILIANO
Università di Pisa Dipartimento di Medicina Clinica e Sperimentale Direttore della Scuola di Specializzazione in Neurologia Clinica Neurologica, Ospedale S. Chiara via Roma 67- 56126 Pisa tel: 050993046; fax: 050554808 E-mail: g.siciliano@med.unipi.it
Proboviro: GIOACCHINO TEDESCHI
Clinica Neurologica II – Seconda Università di Napoli, P.zza Miraglia 2- 80138 Napoli, Tel. 081 5665004, Tel. segreteria/fax 081 5665095, E-mail gioacchino.tedeschi@unicampania.it
• DIRETTIVO STORICO
2023-2025
Presidente: Alessandro Padovani
Vice-Presidente: Vincenzo Andreone
Segretario: Nicola De Stefano
2021-2023
Presidente: Alfredo Berardelli
Vice-Presidente: Rocco Quatrale
Segretario: Antonio Toscano
2019-2021
Presidente: Gioacchino Tedeschi
Vice-Presidente: Massimo Del Sette
Segretario: Alessandro Padovani
2017 – 2019
Presidente: Gianluigi Mancardi
Vice-Presidente: Roberto Eleopra
Segretario: Mario Zappia
2015 – 2017
Presidente: Leandro Provinciali
Vice-Presidente: Elio Clemente Agostoni
Segretario: Leonardo Lopiano
2013 – 2015
Presidente: Aldo Quattrone
Vice-Presidente: Bruno Giometto
Segretario: Carlo Ferrarese
Tesoriere: Mario Zappia
2011 – 2013
Presidente: Giancarlo Comi
Vice-Presidente: Carlo Serrati
Segretario: Alfredo Berardelli
Tesoriere: Gioacchino Tedeschi
2009 – 2011
Presidente: Antonio Federico
Vice Presidente: Giuseppe Micieli
Segretario: Alfredo Berardelli
Tesoriere: Gioacchino Tedeschi
2007-2009
Presidente: Giorgio Bernardi
Vice Presidente: Roberto Sterzi
Segretario: Gioacchino Tedeschi
Tesoriere: Giovanni Luigi Mancardi
2005-2007
Presidente: Mario Manfredi
Vice Presidente: Fabrizio Antonio De Falco
Segretario: Gioacchino Tedeschi
Tesoriere: Giovanni Luigi Mancardi
2003-2005
Presidente: Corrado Messina
Vice Presidente: Tommaso Sacquegna
Segretario: Giovanni Luigi Mancardi
Tesoriere: Leandro Provinciali
2001-2003
Presidente: Nicola Rizzuto
Vice Presidente: Vito Toso
Segretario: Giovanni Luigi Mancardi
Tesoriere: Leandro Provinciali
1999 – 2001
Presidente: Vincenzo Bonavita
Vice Presidente: Bruno Jandolo
Segretario: Leandro Provinciali
Tesoriere: Gianluigi Lenzi
1996-1999
Presidente: Luigi Amaducci † (1996-1997), Bruno Jandolo (1998), Vincenzo Bonavita (1999)
Vice Presidente: Bruno Jandolo
Segretario: Leandro Provinciali
Tesoriere: Gianluigi Lenzi
1993-1996
Presidente: C. Fieschi
Vice Presidente: V. Toso
Segretario: A. Federico
Tesoriere: L.Provinciali
1990-1993
Presidente: M. Carreras; Vice Presidente: F. Cornelio
Segretario: A. Fedecico
Tesoriere: B. Tavolato
1987-1990
Presidente: E. Ferrari
Vice Presidente: C. De Fanti
Segretario: A. Baruzzi
Tesoriere: P. Livrea
1984-1987
Presidente: E. Lugaresi
Vice Presidente: T. Caraceni
Segretario: D. Mancia
Tesoriere: N. Accornero
1982-1984
Presidente: G. A. Buscaino
Vice Presidente: R. Boeri
Segretario: M. Manfredi
Tesoriere: N. Accornero
1980-1982
Presidente: L. Bergamini
Vice Presidente: P. Nicoletti
Segretario: T. Caraceni
Tesoriere: M. Manfredi
1978-1980
Presidente: G. Macchi
Vice Presidente: D. Fontanari
Segretario: L. Amaducci
Tesoriere: M. Manfredi
1976-1978
Presidente: C. Loeb
Vice Presidente: L. Bergamini
Segretario: G. Broggi
Tesoriere: G. Alemà
1974-1976
Presidente: P. Pinelli
Vice Presidente: E. Ferrari
Segretario: R. Boeri
Tesoriere: G. Alemà
1972-1974
Presidente: F. Visintini
Vice Presidente: G. Alemà
Segretario: E. Ferrari
• COMITATO ETICO DI VIGILANZA
GIANLUIGI MANCARDI
E-mail glmancardi@neurologia.unige.it
LEANDRO PROVINCIALI
E-mail leandroprovinciali@gmail.com
MARIA TROJANO
E-mail maria.trojano@uniba.it
• STATUTO
DENOMINAZIONE, SCOPO, SEDE
Art.1 – E’ costituita l’Associazione “Società Italiana di Neurologia (SIN)”. La SIN è Associazione di rilevanza nazionale che accoglie, con le modalità previste nel presente statuto, i medici specialisti e specializzandi che comunque operino in ambito neurologico nelle varie Strutture e settori di attività del Servizio sanitario nazionale (aziende ospedaliere, aziende USL, aziende universitarie, IRCCS, ospedali classificati, case di cura private accreditate, ecc.) o in regime libero-professionale che ne facciano richiesta.
Art.2 – La SIN ha lo scopo di promuovere in Italia gli studi neurologici. Le finalità istituzionali dell’associazione comprendono:
- il miglioramento della qualità professionale nell’ assistenza ai soggetti con malattie del sistema nervoso
- l’attività di aggiornamento professionale e di formazione permanente, residenziale e a distanza, nei confronti degli associati con programmi annuali di attività formativa ECM;
- la collaborazione con il MIUR, le università, il Ministero della salute, le Regioni, le Aziende sanitarie e gli altri organismi e istituzioni sanitarie pubbliche;
- l’elaborazione di linee guida in collaborazione con l’Agenzia per i Servizi Sanitari Regionali (A.S.S.R.) e la F.I.S.M.; promozione di trials di studio e di ricerche scientifiche finalizzate e rapporti di collaborazione con altre società e organismi scientifici.
La SIN non ha fini di lucro ed è espressamente esclusa, anche indirettamente, ogni finalità o attività sindacale. La SIN non potrà esercitare attività imprenditoriali o partecipare ad esse, salvo quelle necessarie per le attività di formazione continua.
Art.3 – La SIN ha sede nel Comune di Siena in Via Del Rastrello n.7. La sede potrà essere in qualunque momento modificata con delibera del Consiglio Direttivo che dovrà essere ratificata dalla prima assemblea.
Art.4 – La SIN può istituire Gruppi di Studio composti da Soci Ordinari interessati ad approfondire particolari aspetti culturali, scientifici e didattici della Neurologia. Può istituire, inoltre, Sezioni Regionali ed Interregionali per promuovere un ampio scambio di informazioni tra i settori speculativi ed applicativi della Neurologia. In analogia con quanto realizzato in altre Società Neurologiche Europee può istituire una Sezione dedicata ai Neurologi in formazione.
Art.5 – E’ previsto il riconoscimento di “Associazione Autonoma Aderente alla SIN” di sodalizi di studiosi impegnati in attività di ricerca, didattica e assistenziale che svolgano un ruolo di giunzione culturale tra la Neurologia Clinica ed altre discipline di base ed applicative.
Il riconoscimento potrà essere deliberato dal Consiglio Direttivo a condizione che sia rispettato quanto stabilito all’art. 25.
Il riconoscimento di “Associazione Autonoma Aderente” alla SIN non comporta alcuna assunzione di responsabilità da parte della SIN come associazione né dei propri Organi o uffici per le obbligazioni assunte dalla ”Associazione Aderente” che conserva ad ogni effetto la più completa autonomia gestionale sia in termini economici che di responsabilità in genere.
Art.6 – La SIN si propone di perseguire gli scopi statutari in particolare mediante:
- l’organizzazione di congressi scientifici nazionali almeno una volta l’anno;
- riunioni scientifiche periodiche delle Sezioni Regionali ed Interregionali;
- altre riunioni programmate e finalizzate alla promozione della ricerca e dell’attività scientifica, alla formazione, anche manageriale dei soci ed alla loro qualità professionale;
- una propria rivista, organo ufficiale della Società;
- un sito SIN e un notiziario SIN quale strumento periodico di informazione;
- il coordinamento delle attività delle Associazioni Autonome Aderenti;
- l’aggiornamento professionale attraverso attività dirette ad adeguare le conoscenze dei soci al progressivo evolvere delle conoscenze neurologiche;
- la concessione di patrocini a iniziative scientifiche e didattiche, purché siano di elevato livello e coerenti con i fini istituzionali della Società. Il patrocinio viene concesso dal Presidente in base alle deliberazioni del Consiglio Direttivo.Il perseguimento delle finalità e la valutazione dei risultati sarà effettuato con adeguati sistemi di verifica del tipo e dellaqualità delle attività svolte.
PATRIMONIO ED ESERCIZI SOCIALI
Art. 7 – Le attività sociali sono finanziate solo attraverso i contributi degli associati e/o di enti pubblici nonché di soggetti privati, con esclusione di finanziamenti che configurino conflitto di interesse con il S.S.N., anche se forniti attraverso soggetti collegati.
Le attività ECM saranno finanziate attraverso l’autofinanziamento e i contributi degli associati e/o enti pubblici e privati, ivi compresi i contributi delle industrie farmaceutiche e di dispositivi medici, nel rispetto, dell’autonomia ed indipendenza dell’Ente e dei criteri e dei limiti stabiliti dalla Commissione nazionale per la formazione continua.
II Patrimonio è costituito:
1) dai beni mobili ed immobili, pervenuti a qualsiasi titolo, che diverranno proprietà della SIN;
2) da eventuali fondi di riserva costituiti dalle liberalità;
3) da eventuali crediti, disponibilità liquide, erogazioni, donazioni e lasciti, pervenuti qualsiasi titolo;
4) da ogni altro bene materiale ed immateriale acquisito con i mezzi della SIN
Le entrate della SIN sono costituite:
a) dalle quote sociali;
b) da liberalità e rimborsi derivanti da manifestazioni culturali e scientifiche o partecipazioni ad esse;
c) dagli utili derivanti dall’attività imprenditoriale necessaria per le attività svolte esclusivamente nell’ambito del Programma nazionale di formazione continua in medicina (ECM);
d) da ogni altra entrata che concorra ad incrementare l’attivo sociale, nel rispetto delle normative vigenti in materia ed in specie relative a finanziamenti che possano configurare conflitto di interesse con il S.S.N. anche se forniti attraverso soggetti collegati;
e) dalle rendite dei beni facenti parte del patrimonio sociale.
Art. 8 – L’esercizio finanziario chiude al 31 dicembre di ogni anno. Entro novanta giorni dalla fine di ogni esercizio verranno predisposti dal Consiglio Direttivo il Bilancio consuntivo e quello preventivo del successivo esercizio.
Tali documenti dovranno essere pubblicati, a cura della Presidenza, nel Sito Istituzionale dell’Ente.
SOCI
Art. 9 – La SIN ha quattro categorie di Soci:
- Soci Ordinari
- Soci Aderenti
- Soci Onorari
- Soci Corrispondenti Esteri
Il domicilio dei soci per ogni rapporto con l’associazione, è quello risultante dall’apposito elenco dei soci. Ove il socio abbia comunicato il proprio indirizzo di posta elettronica, a tale indirizzo potrà essere inviato ogni avviso o comunicazione.
A tal fine dovrà essere annotata sull’elenco dei soci ogni modifica di indirizzo comunicata per scritto dai soci.
Art. 10 – Possono divenire Soci Ordinari, senza limitazioni personali o inerenti il luogo di lavoro, tutti i medici specialisti e specializzandi che comunque operino, anche se non in via esclusiva, in ambito neurologico nelle varie Strutture e settori di attività del Servizio Sanitario Nazionale (aziende ospedaliere, aziende USL, aziende universitarie, IRCCS, ospedali classificati, case di cura private accreditate, ecc.) o in regime libero-professionale che ne facciano richiesta.
La domanda di iscrizione dovrà essere indirizzata al Presidente della SIN. Ad essa dovrà essere allegato un curriculum e tutta la documentazione comprovante il possesso dei requisiti previsti dallo Statuto per l’ammissione L’ammissione è deliberata dal Consiglio Direttivo. Gli ammessi sono tenuti a versare la quota annuale nella misura stabilita ed eventualmente aggiornata dall’Assemblea dei Soci Ordinari.
Essi acquistano diritto di voto in Assemblea sei mesi dopo aver atteso al versamento della quota associativa.
I Soci Ordinari in regola con il pagamento delle quote associative hanno diritto di voto in Assemblea e, senza alcun carico economico aggiuntivo, sarà loro inviata la Rivista, organo ufficiale della SIN.
Art. 11 – Può divenire Socio Aderente chi, pur non rivestendo lo status di Socio Ordinario svolga attività didattica, scientifica e di ricerca che risulti di supporto alle Neuroscienze e partecipi ai lavori dei Gruppi di Studio, delle Sezioni Regionali e Interregionali e delle Associazioni Autonome Aderenti alla SIN.
La qualifica di Socio Aderente può essere acquisita con la semplice presentazione al Consiglio Direttivo della SIN della domanda controfirmata da due soci appartenenti al Gruppo di Studio, alla Sezione o Associazione Autonoma Aderente
Art. 12 – Possono essere riconosciuti Soci Onorari Personalità Italiane e Straniere che si siano particolarmente distinte per la loro attività di studio e di ricerca nell’ambito delle Neuroscienze. La nomina è effettuata con delibera del Consiglio Direttivo con voto favorevole di almeno 3/4 (tre quarti) dei suoi membri
Art. 13 – Possono essere riconosciuti Soci Corrispondenti Esteri, studiosi che svolgano la loro attività in Istituzioni Scientifiche oltre frontiera e che intrattengano rapporti di proficua collaborazione con la SIN.
La nomina è effettuata con delibera del Consiglio Direttivo con voto favorevole di almeno 3/4 (tre quarti) dei suoi membri.
Art. 14 – I Soci Aderenti, i Soci Onorari Italiani e Stranieri ed i Soci Corrispondenti Esteri sono esentati dal pagamento della quota annuale di associazione. Essi non hanno diritto di voto in Assemblea e non possono ricoprire cariche sociali.
Art. 15 – La qualità di Socio in genere si perde per decesso, per dimissione, per indegnità riconosciuta dal Consiglio Direttivo con delibera ratificata dall’Assemblea dei Soci e per morosità nel caso dei Soci Ordinari. La perdita della qualità di Socio esclude ogni rivalsa economica nei riguardi della Società.
ORGANI SOCIALI
Art. 16 – Sono Organi della SIN:
- la Presidenza;
- iI Consiglio Direttivo;
- l’Assemblea dei Soci Ordinari;
- il Collegio dei Probiviri;
E’ espressamente esclusa la retribuzione delle Cariche sociali. Non possono assumere cariche sociali coloro che abbiano subito sentenze di condanna, passate in giudicato, in relazione all’attività dell’Ente. La pronuncia della sentenza di condanna (passata in giudicato) per fatti compiuti in relazione all’attività dell’Ente, costituisce causa di decadenza dalla carica sociale assunta.
PRESIDENZA
Art. 17 – La Presidenza è l’Organo esecutivo della SIN ed è costituita dal Presidente, dal VicePresidente, dal Segretario e dal Tesoriere. Il Presidente, il Vice-Presidente ed il Segretario sono eletti dall’assemblea con votazioni segrete e distinte.
Il Presidente eletto assume le funzioni all’inizio del biennio successivo alla sua elezione.
II Tesoriere viene nominato dal Consiglio Direttivo tra i Consiglieri eletti dall’Assemblea.
Le cariche di Presidente e Vice-Presidente vengono rinnovate ogni due anni e non sono rinnovabili consecutivamente.
Il Presidente rappresenta ufficialmente e giuridicamente la SIN. Egli dovrà avere pertanto una anzianità di iscrizione alla Società e precedenti esperienze gestionali adeguate ai compiti che deve svolgere e dovrà possedere una dimensione culturale ed una produzione scientifica che gli consentano di rappresentare degnamente la Società Italiana di Neurologia nel contesto Nazionale ed Internazionale.
Il Presidente convoca e presiede le Assemblee dei Soci sia Ordinarie che Straordinarie, riunisce il Consiglio Direttivo. Cura che vengano eseguite le delibere del Consiglio Direttivo e le decisioni prese nelle Assemblee, rimanendo costantemente in contatto tramite il Segretario con le Sezioni Regionali e Interregionali e con gli Uffici che fanno capo al Consiglio Direttivo.
Il Presidente e/o il Tesoriere, coerentemente con le deliberazioni del Consiglio Direttivo, sono investiti, anche con firma libera e disgiunta fra loro, dei più ampi poteri per la gestione dei fondi sociali e delle somme liquidate a disposizione della Società, con facoltà di riscuotere somme e valori, di fare pagamenti, di dare e rilasciare quietanze, di provvedere ad operazioni bancarie attive e passive di qualsiasi genere, quali, in via esemplificativa, apertura dei conti correnti, richiesta fidi, anticipazioni, crediti e sovvenzioni e loro utilizzo, emissione di assegni su conti correnti intestati alla Società.
In caso di dimissioni o di impedimento permanente del Presidente in carica, il Vice Presidente ne assume le funzioni fino alla successiva assemblea dei Soci. In tale occasione, con ratifica dell’Assemblea, le funzioni vengono assunte dal Presidente eletto, il quale conclude comunque il proprio mandato alla data prevista all’atto della sua elezione.
Il Segretario, oltre che curare lo svolgimento delle Assemblee e delle sedute del Consiglio Direttivo, delle quali redige i relativi verbali, mantiene uno stretto collegamento con il Presidente, i membri del Consiglio Direttivo, il Tesoriere e gli Uffici che fanno capo al Consiglio Direttivo. Egli inoltre coordina tutte le iniziative idonee alla realizzazione degli scopi statutari della S.I.N.
L’attività del Segretario si avvale della collaborazione di una Segreteria Amministrativa, affidata di norma ad una organizzazione esterna, proposta dal Segretario ed approvata dal Consiglio Direttivo.
II Tesoriere cura insieme al Segretario lo schedario generale dei Soci, controlla il pagamento delle quote ed amministra i beni della SIN.
CONSIGLIO DIRETTIVO
Art. 18 – II Consiglio Direttivo è costituito da quindici consiglieri eletti a scrutinio segreto dall’Assemblea ogni due anni oltre ai componenti di diritto di cui all’ultimo comma del presente articolo. In caso di vacanze, le cariche verranno ricoperte seguendo l’ordine dei voti riportati dai non eletti; a parità di voti risulterà eletto il candidato anagraficamente più anziano.
Le singole candidature per la elezione dei Consiglieri e della Presidenza saranno rese note dal Presidente e l’elenco dei candidati sarà affisso nell’aula in cui si svolgeranno le elezioni. La presentazione delle candidature e le modalità di votazione a scrutinio segreto sono disciplinate da un regolamento elettorale emanato dal Consiglio Direttivo.
Ciascun Socio potrà esprimere un numero di preferenze non superiore ai 2/3 (due terzi) del numero dei consiglieri da eleggere. Risulteranno eletti i quindici candidati che avranno riportato il maggior numero di voti.
I Consiglieri rimangono in carica per un biennio e possono essere rieletti consecutivamente per un altro biennio, in misura non eccedente i 2/3 (due terzi) del numero totale dei consiglieri.
Fanno inoltre parte del Consiglio Direttivo, con diritto di voto, il Presidente uscente, il Presidente eletto, e i Presidenti delle Associazioni Autonome Aderenti alla SIN.
Art. 19 – Il Consiglio Direttivo collabora con la Presidenza per la realizzazione dei fini istituzionali della SIN sulla base di programmi approvati dall’Assemblea coordinando la realizzazione delle iniziative scientifiche e culturali concordate.
Il Consiglio Direttivo può inoltre costituire particolari Commissioni per specifici compiti.
Al Consiglio Direttivo compete inoltre, il riconoscimento delle “Associazioni Autonome Aderenti”.
Il Consiglio Direttivo è convocato dal Presidente per sua iniziativa o su richiesta di almeno 1/4 (un quarto) dei suoi membri o da 1/5 (un quinto) dei Soci Ordinari. Le sedute sono valide qualora sia presente la metà più uno del totale dei membri diminuito del numero degli assenti giustificati dal Presidente.
Il Consiglio Direttivo delibera a maggioranza semplice dei presenti. In caso di parità di voti prevale la proposta cui abbia dato il voto favorevole il Presidente.
Art. 20 – Sono costituiti in seno al Consiglio Direttivo vari Uffici che possono essere affidati anche ad esperti esterni per il raggiungimento dei fini istituzionali della SIN.
II Consiglio Direttivo provvede alla istituzione di un Ufficio per la Rivista, Organo Ufficiale della SIN, costituito dal Redattore Capo e dagli Editori Associati, a cui sono demandate le funzioni di Organo Direttivo, con compiti definiti da un Regolamento formulato o modificato dal Consiglio Direttivo in carica. Tale Ufficio provvederà obbligatoriamente, con le modalità previste dal predetto regolamento, alla pubblicazione e divulgazione dell’attività scientifica attaverso il sito web dell’Ente, aggiornato costantemente.
Il Consiglio Direttivo provvede alla nomina di un Comitato Scientifico per la verifica ed il controllo della qualità delle attività svolte e della produzione tecnico-scientifica, da effetuare secondo gli indici diproduttività scientifica e bibliometrici validati dalla comunità scientifica internazionale.
Il Consiglio Direttivo determinerà il numero dei componeti il Comitato Scientifico, che dovranno essere scelti tra gli associati che si siano distinti per particolari meriti scientifici, con delibera da assumere a maggioranza assoluta.
ASSEMBLEA
Art. 21 -L’Assemblea dei Soci Ordinari deve essere convocata dal Presidente in via ordinaria almeno una volta all’anno ed in concomitanza con le attività scientifiche programmate. Essa elabora e fissa, in confronto concreto con la realtà scientifica, didattica e assistenziale, le linee programmatiche generali della SIN.
Stabilisce inoltre l’ammontare delle quote associative e le modalità di riscossione, approva il Bilancio consuntivo e preventivo e l’attività svolta. L’Assemblea fissa la data e la sede dei congressi nazionali, delle altre attività sociali, i temi e le modalità di svolgimento dei congressi, anche sulla base di proposte presentate dal Consiglio Direttivo.
L’Assemblea, su richiesta anticipata di almeno 60 (sessanta) giorni, può essere convocata in via straordinaria dal Presidente su delibera del Consiglio Direttivo o su richiesta motivata di almeno 1/5 (un quinto) dei Soci Ordinari.
L’Assemblea è l’unico organo competente a modificare lo statuto della SIN o a sciogliere la SIN stessa deliberando su questi argomenti con maggioranza qualificata dei 2/3 (due terzi) dei Soci Ordinari presenti.
In caso di scioglimento della società gli eventuali attivi saranno devoluti, a giudizio della SIN, a favore di enti o associazioni della stessa categoria che perseguono scopi analoghi.
Tutte le deliberazioni dell’Assemblea dei Soci Ordinari entreranno in vigore all’atto dell’approvazione, salvo che diversamente specificato.
Art. 22 – La convocazione, sia ordinaria che straordinaria, dell’Assemblea deve essere fatta almeno un mese prima della data fissata, con avviso spedito all’indirizzo di posta elettronica comunicato nonchè tramite pubblicazione dell’avviso di convocazione sulla rivista della Sin (Neurological Sciences) e sul sito della Sin (www.neuro.it).
L’Assemblea è valida in prima convocazione se è presente almeno la metà dei Soci Ordinari, in seconda convocazione (che può avvenire anche nella stessa giornata e trascorsa un’ora da quella fissata per la prima convocazione) se è presente almeno 1/10 (un decimo) dei Soci Ordinari. Le deliberazioni sono adottate a maggioranza semplice dei presenti, salvo i casi di cui al precedente articolo 21.
L’Assemblea verrà presieduta dal Presidente della Associazione, o, in sua assenza, dal Vice Presidente o, in caso di assenza di entrambi, dalla persona eletta dall’Assemblea A cura del Segretario – o in sua assenza – di altro consigliere designato dal Presidente dell’Assemblea – verrà steso un verbale dell’Assemblea, che dovrà essere firmato dal Presidente dell’Assemblea stessa e da chi lo ha redatto. Ogni socio ha diritto di prendere visione del verbale che verrà conservato agli atti.
I soci morosi non possono esercitare il diritto di voto e pertanto non sono calcolati ai fini dei quorum costitutivi e deliberativi.
COLLEGIO DEI PROBIVIRI
Art. 23 – Il Collegio dei Probiviri è costituito da tre Soci Ordinari, iscritti alla Società da almeno dieci anni, eletti dall’Assemblea dei Soci nella stessa seduta in cui viene eletto il Consiglio Direttivo. Essi sono rieleggibili consecutivamente per non oltre due mandati. I tre Probiviri verranno eletti dall’Assemblea su scheda separata.
Ciascun socio potrà esprimere un numero di preferenze non superiore ai 2/3 (due terzi) del numero totale da eleggere.
Il Collegio elegge fra i suoi membri un Presidente.
Il Collegio dei Probiviri è convocato dal Presidente ogni qualvolta sia richiesta una sua deliberazione. Le sedute sono valide qualora siano presenti tutti i membri in carica.
Il Collegio dei Probiviri delibera a maggioranza semplice dei presenti.
Il Collegio dei Probiviri esprime parere consultivo, su richiesta del Consiglio Direttivo, in merito alle attività espletate dai Soci in nome o per conto della Società, tenendo conto dei principi di tutela della Società, dei suoi componenti e degli aspetti pertinenti l’attività societari.
Tutte le eventuali controversie sociali tra Soci e tra questi e la SIN o suoi Organi, saranno sottoposte, con esclusione di ogni altra giurisdizione, alla competenza del Collegio dei Probiviri; essi giudicheranno ex bono et aequo senza formalità di procedura. II loro lodo sarà inappellabile.
SEZIONI REGIONALI – SEZIONI INTERREGIONALI – SEZIONE DEI NEUROLOGI IN FORMAZIONE -GRUPPI DI STUDIO
Art. 24 – Le Sezioni Regionali ed Interregionali hanno una propria Assemblea ed un Segretario, coadiuvato da un Collegio di Segreteria, la cui attività è disciplinata da apposito regolamento. Eventuali modalità organizzative difformi da quelle previste in questo regolamento dovranno essere preliminarmente autorizzate dal Consiglio Direttivo SIN.
La Sezione dei Neurologi in formazione consente l’afferenza alla SIN dei giovani cultori delle scienze neurologiche a condizioni agevolate. Essa elegge i propri rappresentanti destinati anche alla partecipazione alle riunioni internazionali con analoghe Sezioni di Società Neurologiche straniere
Art. 25 – I Gruppi di Studio, su loro richiesta, potranno essere riconosciuti come “Associazioni Autonome Aderenti” alla SIN alle seguenti condizioni:
– che lo Statuto della costituenda Associazione venga preventivamente approvato dal Consiglio Direttivo della SIN e ratificato dall’Assemblea dei Soci Ordinari;
– che lo Statuto della costituenda Associazione preveda la partecipazione del Presidente della SIN al proprio Consiglio Direttivo con diritto di voto;
– che in sede di costituzione, la nuova Associazione conti non meno di quindici Soci fondatori;
– che il Gruppo di Studio e quindi la costituenda associazione, abbiano le caratteristiche previste dall’art.5 di questo statuto.
• CODICE ETICO
I codici di condotta sono delle regole di autodisciplina che un determinato settore della società dichiara di impegnarsi a seguire nello svolgimento del proprio ruolo, rispecchiando particolari criteri di adeguatezza e opportunità, in riferimento a un determinato contesto culturale, sociale o professionale.
Fanno parte di quelle iniziative volontarie nate sull’onda delle teorie di alcuni economisti che negli anni ‘80 hanno iniziato a parlare di responsabilità sociale delle società: una visione di società sostenibile che ha prodotto alcuni strumenti che aiutano a realizzare questi obiettivi. L’adozione di codici etici di autoregolazione o codici di condotta è uno di questi strumenti che mirano a rendere pubblica la responsabilità sociale della società (Corporate Social Responsibility).
Un codice di condotta rappresenta quindi un contratto sociale tra la società e i suoi stakeholders e ha la funzione di legittimarne l’autonomia annunciando pubblicamente che essa è consapevole dei suoi obblighi di cittadinanza, che ha sviluppato politiche e pratiche societarie coerenti con questi obblighi e che teoricamente è in grado di attuarle attraverso appropriate strutture organizzative e sanzioni.
I codici di condotta nelle società
Il codice di condotta di una società può definirsi come la Carta Costituzionale della società, una carta dei diritti e doveri morali che definisce la responsabilità etico-sociale di ogni partecipante all’organizzazione. E’ un mezzo a disposizione delle società per prevenire comportamenti irresponsabili o illeciti da parte di chi opera in nome e per conto della società, perché introduce una definizione chiara ed esplicita delle responsabilità etiche e sociali dei propri rappresentanti, dipendenti e spesso anche fornitori, verso i diversi gruppi di interesse. E’ considerato il principale strumento di implementazione dell’etica all’interno della società ma anche un mezzo per sostenerne la reputazione, in modo da creare fiducia verso l’esterno.
Il codice di condotta è dunque uno strumento di gestione con cui la società tende a esplicitare i tratti essenziali della propria identità attuale e futura meglio conosciute come mission e vision.
• CODICE CONDOTTA PROFESSIONALE
Informiamo tutti i soci SIN che la Società Italiana di Neurologia si è dotata del Codice di Condotta Professionale del neurologo, disponibile e visionabile al seguente link:
• GENDER EQUALITY PLAN
Nel novembre 2023 è stata costituita la consulta di genere nell’ambito del direttivo della SIN che ha sviluppato un Gender Equality Plan (GEP), pubblicato nel marzo 2024, con l’obiettivo di identificare la strategia per l’uguaglianza di genere nella SIN.
• SIN SDGs
LA SIN e I Sustainable Development Goals
I Sustainable Development Goals (SDGs) costituiscono un insieme di obiettivi globali adottati dalle Nazioni Unite per affronte le side più urgenti del nostro tempo, come la povertà, l’ineguaglianza e il cambiamento climatico.
Questi obiettivi rappresentano un’importante opportunità per le società scientifiche di contribuire a un futuro sostenibile e prospero.
A nome del presidente SIN, prof. Alessandro Padovani, Vi invitiamo a prenderne visione e a riflettere su come possono essere integrati nelle strategie e attività quotidiane.
CLICCA QUI PER SCARICARE IL DOCUMENTO
• VALUTAZIONE DI IMPATTO SULLA PROTEZIONE DEI DATI
La DPIA, acronimo di Data Protection Impact Assessment, è una valutazione preliminare, eseguita dal titolare del trattamento dei dati personali, relativa agli impatti a cui andrebbe incontro un trattamento laddove dovessero essere violate le misure di protezione dei dati.
In linea con l’approccio basato sul rischio adottato dal regolamento generale sulla protezione dei dati, non è obbligatorio svolgere una valutazione d’impatto sulla protezione dei dati per ciascun trattamento; è necessario realizzare una valutazione d’impatto sulla protezione dei dati soltanto quando la tipologia di trattamento “può presentare un rischio elevato per i diritti e le libertà delle persone fisiche” (articolo 35 paragrafo 3 del Regolamento 2016/679).
Clicca qui per scaricare il documento completo.
• DOC VALUTAZIONE RISCHIO BIOLOGICO IN EVENTO (DVRE)
Per la pianificazione e realizzazione di eventi in sicurezza, FONDAZIONE SIN adotta questo documento di valutazione del rischio (DVRE) redatto dal Gruppo di Lavoro di Federcongressi&eventi appositamente costituito e partecipato da rappresentanti di diverse categorie di operatori che costituiscono la filiera della Meeting Industry italiana (organizzatori di eventi, provider ECM, caterer, fornitori di servizi, etc).
Il documento, per sua natura, è soggetto a revisioni continue che saranno basate sull’evoluzione normativa e sulle segnalazioni di aspetti specifici, criticità e problematiche particolari che potranno essere rilevate dai soci di Federcongressi&eventi grazie alla loro esperienza sul campo. Le segnalazioni saranno inviate alla Segreteria nazionale dell’associazione all’indirizzo federcongressi@federcongressi.it.
E’ possibile visionare il Documento cliccando su: Documento di Valutazione del Rischio biologico in Evento (DVRE)
• MOG
Per evitare le gravi conseguenze che derivano agli enti (società di capitali, società di persone, associazioni, ecc.) dalla commissione di un reato da parte di un rappresentante e/o di un dipendente, la Società Italiana di Neurologia ha deciso di dotarsi di un modello di organizzazione e gestione.
Tra le sanzioni più gravi vi sono: l’interdizione dall’esercizio dell’attività; l’esclusione da agevolazioni, finanziamenti, contributi o sussidi e l’eventuale revoca di quelli già concessi; il divieto di contrattare con la pubblica amministrazione; la sospensione o la revoca delle autorizzazioni, licenze o concessioni funzionali alla commissione dell’illecito, ecc.
Il modello organizzativo impedisce – quando efficacemente predisposto ed attuato – l’applicazione di sanzioni pecuniarie ed interdittive a carico della società.
• UNI EN ISO 9001:2015
L’Ufficio di Presidenza della SIN ha deciso di applicare un Sistema di Gestione per la Qualità, in accordo ai requisiti della norma UNI ENI ISO 9001 edizione settembre 2015, avendo individuato nell’adozione dei principi dell’Assicurazione della Qualità la migliore scelta per il miglioramento dell’efficenza della SIN: ciò al fine di individuare e soddisfare sempre meglio le esigenze dei clienti in un’ottica di miglioramento continuo del prodotto, del servizio e dei processi, nonchè di consolidamento ed ampliamento della posizione dell’azienda all’interno del mercato di riferimento
• REGOLAMENTO ELETTORALE
REGOLAMENTO ELETTORALE SIN
- Ciascuna candidatura dovrà essere accompagnata da un breve profilo del candidato (non più di 15-20 righe), che individui il ruolo professionale, precedenti incarichi societari e campi di interesse. A norma di Statuto i componenti il Collegio dei Probiviri dovranno essere iscritti alla Società da almeno 10 anni ed il candidato a Presidente dovrà avere le caratteristiche previste dall’art. 17 dello Statuto;
- Ciascuna candidatura sarà inserita sul sito web della SIN;
- Il Consiglio Direttivo nominerà nella prossima riunione utile un Comitato Elettorale formato da 5 soci che presiederanno alle votazioni ed allo spoglio delle schede elettorali;
- I risultati saranno comunicati il giorno successivo le votazioni, durante l’Assemblea dei Soci SIN;
- Si rammenta che ciascun socio non potrà esprimere più di 10 preferenze per l’elezione dei 15 Consiglieri e più di 2 preferenze per l’elezione dei 3 componenti il Collegio dei Probiviri;
- Come da art. 10 dello Statuto i soci, approvati nell’anno corrente, acquistano diritto di voto in Assemblea, sei mesi dopo aver atteso al versamento della quota associativa;
Occorre comunque, benchè non sancito da alcuna norma attualmente in vigore, che i candidati siano in regola con il pagamento delle quote associative annuali e che sia garantita una buona rappresentanza di entrambi i generi nelle candidature.
• REGOLAMENTO SEZIONI REGIONALI
• REGOLAMENTO GDS
• REGOLAMENTO ASSOCIAZIONI
(Approvato nella riunione del Consiglio Direttivo della Società Italiana di Neurologia del 08.05.2019)
- La Società Italiana di Neurologia (SIN) ha lo scopo di favorire e promuovere lo sviluppo della ricerca scientifica e di migliorare la formazione e la qualità professionale degli specialisti in Neurologia.
- A tal fine la SIN incoraggia e sostiene l’aggregazione di associazioni autonome aderenti come recitano l’art. 5 e 25 dello statuto SIN attualmente in vigore:
(ex Art.5) – È previsto il riconoscimento di “Associazione Autonoma Aderente alla SIN” di sodalizi di studiosi impegnati in attività di ricerca, didattica e assistenziale che svolgano un ruolo di giunzione culturale tra la Neurologia Clinica ed altre discipline di base ed applicative.
Il riconoscimento potrà essere deliberato dal Consiglio Direttivo a condizione che sia rispettato quanto stabilito all’art. 25.
Il riconoscimento di “Associazione Autonoma Aderente” alla SIN non comporta alcuna assunzione di responsabilità da parte della SIN come associazione né dei propri Organi o uffici per le obbligazioni assunte dall’”Associazione Aderente” che conserva ad ogni effetto la più completa autonomia gestionale sia in termini economici che di responsabilità in genere.
(ex Art.25) – I Gruppi di Studio, su loro richiesta, potranno essere riconosciuti come “Associazioni Autonome Aderenti” alla SIN alle seguenti condizioni:
– che lo Statuto della costituenda Associazione venga preventivamente approvato dal Consiglio Direttivo della SIN e ratificato dall’Assemblea dei Soci Ordinari;
– che lo Statuto della costituenda Associazione preveda la partecipazione del Presidente della SIN al proprio Consiglio Direttivo con diritto di voto;
– che in sede di costituzione, la nuova Associazione conti non meno di quindici Soci fondatori;
– che il Gruppo di Studio e quindi la costituenda associazione, abbiano le caratteristiche previste dall’art.5 di questo statuto.
L’articolo 25 fa riferimento alla trasformazione di Gruppi di Studio SIN in associazioni e pertanto si usa l’espressione “costituenda” ma tali requisiti si intendono applicabili anche ad associazioni / società già costituite. - Potranno proporre domanda di aderenza le associazioni / società che operino nel campo medico scientifico della neurologia e delle neuroscienze, ne perseguano fini di ricerca e /o aggiornamento professionale, svolgendo attività scientifica e culturale. All’atto della richiesta di adesione, l’associazione / società che intende aderire a SIN deve fornire proprio statuto, atto costitutivo, eventuale logo ufficiale.
- Lo statuto dell’associazione / società richiedente deve essere redatto in forma pubblica e conforme alla normativa vigente, con espressa indicazione della denominazione dell’ente, patrimonio e sede sociale.
- L’associazione / società richiedente deve garantire al proprio interno organi democraticamente eletti con votazione a scrutinio segreto e con durata limitata nel tempo e regolamentazione degli organi associativi e loro modalità di delibera; norme relative all’estinzione societaria e devoluzione del patrimonio; inoltre deve garantire la presenza del presidente SIN alle riunioni del proprio Consiglio Direttivo con diritto di voto, analogamente e con assoluto diritto di reciprocità, a quanto assicurato da SIN per il presidente dell’associazione / società richiedente.
- L’associazione / società richiedente deve dimostrare di avere svolto nell’ultimo triennio della sua esistenza una valida attività scientifica e di ricerca, a insindacabile giudizio del Consiglio Direttivo SIN.
- L’associazione / società richiedente deve documentare il numero dei propri aderenti, che comunque non potrà essere inferiore a 80 soci effettivi.
- L’associazione / società richiedente deve garantire di avere autonoma situazione economica, e come anche citato nell’art. 5 dello statuto SIN, sarà la stessa associazione / società richiedente a esercitare completa autonomia gestionale sia in termini economici che di responsabilità. Deve altresì garantire attività economiche trasparenti e bilanci approvati dall’assemblea annualmente, ove applicabile.
- La SIN, pur favorendo in generale la spontanea formazione e aggregazione di studiosi, non può consentire una eccessiva frammentazione e parcellizzazione delle diverse aggregazioni di soci o metodiche di indagine delle malattie neurologiche. Le associazioni / società richiedenti devono essere portatrici di argomenti che rappresentino un interesse generale e che coinvolgano l’attività di un numero di soci sufficientemente ampio.
- Al fine di regolamentare le richieste di adesione, in analogia a quanto già accade per i Gruppi di Studio SIN, la SIN istituisce una Commissione istruttoria formata dal Presidente, dai Probiviri e dal Referente per la Ricerca SIN, con lo scopo di elaborare e formalizzare le diverse richieste di adesione. La Commissione porterà una relazione motivata sulle singole richieste all’attenzione della Presidenza, che esprimerà un suo giudizio sulla opportunità di accettare la domanda di adesione, presentandolo alla discussione e alla approvazione in occasione della prima riunione utile del Consiglio Direttivo SIN.
- L’associazione / società richiedente è ammessa a titolo provvisorio dopo l’accettazione da parte del Consiglio Direttivo SIN. La piena adesione potrà avvenire solo dopo la ratifica da parte dell’assemblea dei soci SIN.
- La SIN ritiene di primaria importanza che le conoscenze e gli avanzamenti prodotti dalle diverse associazioni / società aderenti vengano diffusi a tutti i soci della Società e non restino confinati nell’ambito delle singole realtà. Pertanto dovranno essere messe in opera tutte le iniziative necessarie alla divulgazione delle attività svolte: a questo proposito SIN garantisce a ciascuna associazione / società aderente visibilità e presenza all’interno del proprio congresso nazionale annuale.
- La qualifica di associazione / società autonoma si perde per recesso volontario di una delle parti, per modifica dell’oggetto e degli scopi della propria attività, o se questi si rivelassero contrari ai principi dell’ordinamento giuridico vigente.
• REGOLAMENTO CONGRESSO SIN
• RICHIESTA PATROCINIO
Come richiedere il Patrocinio Sin
Le richieste di patrocinio dovranno essere segnalate nella pagina ATTIVITA’ FORMATIVA, seguendo le seguenti indicazioni richieste almeno tre mesi prima dell’evento.
”Una volta effettuata la registrazione sul sito www.neuro.it , sarà possibile accedere nella pagina “attività formativa” e iniziare la procedura di richiesta di patrocinio cliccando su “Segnala Congresso”.
Vi preghiamo di compilare tutti i campi richiesti e di selezionare nell’area “dettaglio” nella sezione “Tipo organizzatore”: Altro e spuntare la Richiesta di patrocinio.”
Si prega di leggere con attenzione il nuovo regolamento adottato dalla Società di Neurologia, contenente il format per la richiesta di patrocinio che andrà allegato alla richiesta online.
I richiedenti che avranno ottenuto il patrocinio riceveranno una comunicazione scritta ufficiale dell’avvenuta concessione e la relativa informazione sarà pubblicata sul sito web SIN nel link riservato ai Patrocini.
Per ogni altra eventuale informazione, si prega di contattare la Segreteria SIN info@neuro.it Tel. 0577 286003
• BILANCI
• PRESENTAZIONE
Schede di patologia
Presentazione
La Società Italiana di Neurologia (Sin) annovera fra i suoi compiti istituzionali non soltanto quello di favorire la ricerca e la formazione dei neurologi ma anche quello di adoperarsi per migliorare la cura e l’ assistenza alle persone che soffrono a causa di malattie del sistema nervoso.
Le patologie neurologiche sono numerose e frequenti e annoverano :
- malattie cerebrovascolari, come l’ictus,
- malattie degenerative come il decadimento mentale, la malattia di Parkinson, i Parkinsonismi, la sclerosi laterale amiotrofica e le atassie cerebellari,
- malattie infiammatorie immuno-mediate come la sclerosi multipla,
- l’epilessia,
- i disturbi del sonno,
- le malattie del sistema nervoso periferico e muscolare,
- le cefalee,
- i tumori cerebrali
- i traumi cranici
oltre alle numerose e complesse malattie neurologiche rare che molto frequentemente colpiscono il sistema nervoso.
La Società Italiana di Neurologia SIN ha deciso di aprirsi al mondo esterno, offrendo le proprie conoscenze e l’ampio sapere dei suoi 3000 neurologi iscritti e dedicati alle piu’ diverse patologie, alle persone che desiderino ricevere informazioni e ambiscano a migliorare le proprie conoscenze sulle malattie neurologiche.
A questo proposito abbiamo redatto alcune schede di malattia pubblicate su questo sito web www.neuro.it dove, chi lo desidera, potrà consultare informazioni semplici, brevi, precise e elaborate dai più importanti studiosi italiani della materia.
Abbiamo reso pubblico sul sito Sin anche l’elenco delle Unità Operative Complesse di Neurologia italiane, a cui ci si potrà rivolgere direttamente per ricevere assistenza e informazioni dirette.
Nell’ottica di una sempre maggiore sinergia con le persone interessate e /o coinvolte da malattie del sistema nervoso, abbiamo anche aperto uno sportello pubblico virtuale intitolato: “L’esperto risponde”.
In questa rubrica un neurologo Sin, particolarmente competente su una determinata patologia, risponderà a specifiche domande rivoltegli dal pubblico.
Riteniamo si tratti di un obiettivo culturale di vasta portata, che desidera contribuire al miglioramento delle conoscenze sulle malattie neurologiche, fornendo risposte precise e scientificamente aggiornate alle persone interessate da queste patologie, ma anche a colleghi o a chiunque sia interessato ai problemi di cui ci occupiamo.
Desideriamo infine fare sapere a tutti coloro che si interessano o che soffrono di malattie neurologiche che esiste una comunità di specialisti molto attiva che si occupa dei loro problemi, e non solo studiano, ricercano e insegnano la materia ai colleghi più giovani, ma anche hanno un rapporto diretto e sinergico con coloro che quotidianamente devono ogni giorno confrontarsi con questi importanti problemi.
Prof Gianluigi Mancardi,
Presidente Sin
• CEFALEE
Schede di patologia
Cefalee
Il capitolo cefalee è di rilevante importanza nell’ambito delle Neuroscienze sia per gli aspetti clinici di diagnostica differenziale, sia per la ricerca. Le cefalee rappresentano un sintomo importante in molte condizioni patologiche del sistema nervoso centrale e in questo caso vengono definite cefalee secondarie.
Esistono cefalee che sono esse stesse la malattia e vengono definite cefalee primarie.
La Classificazione Internazionale delle Cefalee adotta una struttura classificativa di tipo gerarchico in cui tutte le cefalee vengo sistematizzate in livelli diagnostici suddivisi in gruppi principali (primo livello diagnostico), a loro volta suddivisi in livelli successivi (livello 2 o 3), che codificano i diversi tipi e sottotipi, in relazione alle caratteristiche cliniche (numero di attacchi, durata, intensità, presenza della fase algica, sintomi associati) e alla frequenza.
Viene in genere richiesta una diagnosi al primo e secondo livello; mentre è di competenza dello specialista in materia riconoscere le entità codificate ai livelli diagnostici superiori.
Al primo livello di diagnosi si distinguono 14 differenti gruppi:
– dall’ 1 al 4 le cefalee primarie così definite in quanto non riconoscono una causa patologica sottostante: emicrania, cefalea di tipo tensivo, cefalea a grappolo e altre cefalee autonomiche trigeminali e altre forme di cefalee primarie;
– dal 5 al 12 le cefalee secondarie, attribuite ad una specifica noxa patogena o a un particolare disturbo;
– il 13 e il 14 le neuropatie dolorose craniche ed altri dolori facciali.
CEFALEE PRIMARIE
Vengono trattate in dettaglio le due forme più frequenti, l’emicrania e la cefalea di tipo tensivo.
1.Emicrania
- Emicrania senz’aura
- Emicrania con aura
- Emicrania cronica
- Complicanze dell’emicrania
- Emicrania probabile
- Sindromi episodiche che possono essere associate
Epidemiologia
E’ presente nel 14,4% della popolazione mondiale, con una prevalenza 3 volte maggiore nelle donne (18,9%) rispetto agli uomini (6,8%). In Italia prevale in circa il 25% dei soggetti di cui il 32,9% di sesso femminile e il 13% di sesso maschile.
Ha un carattere ciclico per la ricorrenza degli attacchi emicranici che possono avere un andamento episodico, ma anche cronico per la presenza di più di 15 giorni di cefalea al mese.
Patogenesi
E’ una condizione patologica complessa che vede coinvolte strutture craniche algosensibili in uno “stato” cerebrale di alterata eccitabilità e disfunzione dei meccanismi centrali che controllano il dolore, in grado di attivare il sistema trigemino-vascolare in soggetti geneticamente predisposti.
Vengono ipotizzate due teorie fisiopatogenetiche.
La teoria vascolare in cui la vasodilatazione a livello dei vasi meningei rilascia neuropeptidi e sostanze proinfiammatorie capaci di determinare la sensitivizzazione dei neuroni periferico e centrale, del sistema trigemino-vascolare.
La teoria neurogenica dovuta all’attivazione del sistema trigemino vascolare con interessamento del nervo trigemino e le sue fibre nervose che innervano i vasi meningei ed altre strutture del troncoencefalo.
Il rilascio del neuropeptide CGRP (Calcitonine Gene Related Peptide), potente vasodilatatore i cui recettori sono ampiamente diffusi sia perifericamente (ganglio trigeminale, nucleo trigeminale caudale, C1-C2) che centralmente (tronco encefalo, talamo), riveste un ruolo rilevante nella fisiopatogenesi dell’emicrania per lo sviluppo di farmaci che ne blocchino il rilascio.
Diagnosi e Sintomi
La Classificazione Internazionale fornisce una serie di criteri diagnostici e caratteristiche cliniche che devono essere soddisfatti per ogni forma di cefalea raccolti in serie, contrassegnate da lettere dell’alfabeto.
Viene contemplata anche la forma “probabile” quando non vengono rispettati i criteri diagnostici attribuiti alle forme certe, tranne uno.
Sono descritti i due sottotipi di emicrania più frequenti.
Emicrania senz’aura
- Almeno 5 attacchi che soddisfano i criteri B-D
- La cefalea dura 4-72 ore (non trattata o trattata senza successo)
- La cefalea presenta almeno due delle seguenti caratteristiche:
- localizzazione unilaterale
- dolore di tipo pulsante
- dolore con intensità media o forte
- aggravata da che limiti le attività fisiche di routine (per es. camminare, salire
scale)
- Alla cefalea si associa almeno una delle seguenti condizioni:
- presenza di nausea e/o vomito
- presenza di fotofobia e fonofobia
- Non meglio inquadrata da altra diagnosi
Emicrania con aura
- Almeno 2 attacchi che soddisfano i criteri B-C
- Uno o più dei seguenti sintomi dell’aura completamente reversibili
- visivi
- sensitivi
- parola/linguaggio
- motori
- del tronco encefalo
- retinici
- Almeno due delle quattro seguenti caratteristiche:
- almeno un sintomo dell’aura si sviluppa gradualmente in > 5 minuti e/o due o
più sintomi si verificano in successione
- ogni singolo sintomo dura 5-60 minuti
- almeno un sintomo dell’aura è unilaterale
- l’aura è accompagnata o seguita entro 60 minuti da cefalea
- Non meglio inquadrata da altra diagnosi
Terapia
Terapia dell’attacco: triptani, FANS e analgesici; antiemetici.
Terapia di profilassi: beta-bloccanti; calcio-antagonisti; inibitori dell’angiotensina; antidepressivi triciclici; neuro modulatori; anticorpi monoclonali anti-CGRP; nutraceutici; Tossina botulinica di tipo A.
Terapie non farmacologiche: agopuntura, terapie cognitivo comportamentali
- Cefalea di tipo tensivo (CTT)
2.1 Cefalea di tipo tensivo episodica sporadica
2.2 Cefalea di tipo tensivo episodica frequente
2.3 Cefalea di tipo tensivo cronica
2.4 Probabile cefalea di tipo tensivo
Epidemiologia
La CTT è tra le più comuni forme di cefalee primarie, con una prevalenza nella popolazione generale tra il 46% e il 78%, lievemente maggiore nel sesso femminile. Si manifesta a tutte le età, ma più frequentemente prima dei 40 anni.
Patogenesi
L’eziologia non è chiara, ma si pensa che sia multifattoriale e coinvolga aspetti genetici ed ambientali. Situazioni locali anatomiche o parafisiologiche quali determinate posture che mantengono per molte ore la muscolatura del collo o della colonna cervicale o delle spalle in tensione, anomalie della masticazione favorirebbero un imput nocicettivo dalla periferia a strutture centrali facilitandone la percezione del dolore a livello limbico o alterazione delle vie discendenti di modulazione della nocicezione.
Diagnosi e Sintomi
- Almeno 10 attacchi che si verificano in media >1 giorno al mese (
- Durata 30 minuti a 7 giorni
- Almeno due delle seguenti quattro caratteristiche:
- localizzazione bilaterale
- qualità gravativa o costrittiva (non pulsante)
- intensità lieve o media
- non aggravata da che limiti le attività fisiche di routine (per es. camminare, salire scale)
- Si verificano entrambe le seguenti condizioni:
- assenza di nausea e/o vomito
- può essere presente fotofobia oppure fonofobia ma non entrambe
- Non meglio inquadrata da altra diagnosi.
Terapia
Terapia dell’attacco: FANS e paracetamolo; miorilassanti .
Terapia di profilassi: antidepressivi triciclici ; miorilassanti; neuromodulatori nutraceutici.
Terapie non farmacologiche: terapie cognitivo comportamentali (biofeedback); relaxation training.
- Cefalea a grappolo e altre cefalee autonomiche trigeminali
Sono forme rare e soprattutto dell’età adulta.
La caratteristica comune di questo gruppo di cefalee è la presenza della componente autonomica in relazione all’attivazione del sistema parasimpatico, caratterizzata da ptosi, miosi, iniezione congiuntivale, lacrimazione, congestione nasale e rinorrea, edema palpebrale, arrossamento e sudorazione, singolarmente o in combinazione tra loro, sempre localizzati dal lato del dolore; dalla brevità degli attacchi (tranne per la forma continua) e dal periodismo tipico della forma principale di questo gruppo, la cefalea a grappolo, che deve il suo nome all’andamento temporale degli attacchi.
3.1 Cefalea a grappolo
3.2 Emicrania parossistica
3.4 Shurt Lasting Unilateral Neuralgiform headache attacks with Conjunctival injection and Tearing (SUNCT)
3.5 Emicrania continua
3.6 Probabile cefalea autonomico-trigeminale
- Altre cefalee primarie
- Cefalea primaria da tosse
- Cefalea primaria da attività fisica
- Cefalea primaria associata ad attività sessuale
- Cefalea primaria “a rombo di tuono”
- Cefalea da stimolo freddo
- Cefalea da pressione esterna
- Cefalea primaria trafittiva
- Cefalea nummulare
- Cefalea ipnica
- New Daily Persistent Headache (NDPH)
CEFALEE SECONDARIE
Le cefalee secondarie in quanto “cefalee di nuova insorgenza” si verificano in stretta correlazione temporale con un evento che è noto essere causa di cefalea.
Rappresentano il 20% di tutte le forme di cefalea.
I criteri diagnostici prevedono la dimostrazione di una condizione o di una patologia che deve manifestarsi in stretta relazione con la cefalea che migliora o regredisce entro 3 mesi o per periodi minori, dall’eliminazione o dalla remissione spontanea del fattore causale.
Distinguiamo cefalee attribuite a traumi del capo o del collo, a disordini vascolari cranici o cervicali, a disordini intracranici non vascolari, a sostanze o alla loro sospensione, a infezioni craniche, a disordini dell’omeostasi, a disordini di pertinenza oftalmologica, ORL, a disordini psichiatrici.
NEVRALGIE CRANICHE
Le nevralgie craniche includono un insieme di condizioni dolorose parossistiche del capo di riscontro poco frequente nella pratica clinica.
Differenziabili in relazione del nervo interessato e quindi in base allo specifico territorio cutaneo di innervazione, sede del dolore, condividono alcuni aspetti caratteristici: unilateralità del dolore, la manifestazione in attacchi dolorosi brevi e ripetuti, la qualità superficiale e a “scossa elettrica” della sensazione dolorosa e la specifica modalità di scatenamento.
Rientrano in questo capitolo:
- nevralgia del trigemino
- nevralgia del glossofaringeo
- nevralgia del nervo intermedio
- nevralgia occipitale
- neurite ottica
- cefalea da paralisi ischemica dell’oculomozione
- sindrome di Tolosa.Hunt
- sindrome di Raeder
- neuropatia oftalmoplegica dolorosa ricorrente
- sindrome della bocca bruciante
- dolore facciale persistente idiopatico
dolore neuropatico centrale
• DEMENZE
Schede di patologia
Demenze
Il termine ‘demenza’ indica in generale una condizione caratterizzata da deficit della sfera cognitiva (attenzione, memoria, linguaggio, orientamento, ragionamento, critica, personalità e comportamento), che siano: 1. multipli (es. un paziente che in seguito ad un ictus sviluppa un puro problema di linguaggio non viene definito demente), 2. sufficientemente gravi da ridurre l’autonomia del paziente nello svolgimento delle attività quotidiane, 3. presenti stabilmente, non solo in momenti particolari come durante una malattia o un forte stress, e 4. riguardanti funzioni che in precedenza erano del tutto nella norma (es. un paziente con ritardo mentale dalla nascita non viene definito demente).
Negli ultimi decenni, al concetto di demenza si è affiancato quelli di ‘deterioramento cognitivo lieve’ (Mild Cognitive Impairment, MCI), che in oltre il 50% dei casi rappresenta una fase di pre-demenza: tali pazienti presentano inizialmente deficit cognitivi lievi, singoli (es. solo compromissione della memoria) o multipli, che non limitano la loro indipendenza funzionale, ma possono poi progredire a demenza vera e propria. Si tratta di una condizione importante da individuare perchè permette di agire con interventi preventivi che ritardino il passaggio a demenza.
Esistono varie forme di demenza, che hanno cause e sintomi diversi, ma le due tipologie principali sono rappresentate dalle demenze neurodegenerative, che comprendono, tra le altre, la malattia di Alzheimer (MA), la demenza a corpi di Lewy e la patologie fronto-temporali, e dalla demenza di origine vascolare.
Epidemiologia
Una delle più recenti stime di prevalenza della demenza nel mondo riferisce di circa 50 milioni di soggetti affetti. Un valore destinato a raggiungere i 130 milioni entro i prossimi trent’anni. Fino al 60% di questi pazienti hanno una MA, che è la forma di demenza più frequente in assoluto. Attualmente in Italia si stima che siano 600.000 i malati di Alzheimer, corrispondenti a circa il 20% della popolazione ultrasessantenne, ed in una percentuale simile di casi è presente una condizione di MCI. Altre forme neurodegenerative, quali la malattia a corpi di Lewy o le demenze fronto-temporali, rappresentano invece nel loro insieme meno del 20% delle demenze. La demenza vascolare, invece, è la seconda forma più comune di demenza dopo la MA, con una prevalenza stimata, in Europa, compresa tra il 20% e il 40%.
Uno dei principali fattori di rischio per lo sviluppo di una demenza è l’età anziana. Per la MA, in particolare, l’incidenza aumenta in maniera esponenziale con l’avanzare dell’età, passando da quattro nuove diagnosi all’anno ogni 1000 persone tra i 60 ed i 64 anni, a 105 casi /anno/1000 persone negli ultranovantenni. Ulteriori fattori di rischio sono patologie come il diabete, l’ipercolesterolemia, l’ipertensione e l’obesità, ed anche uno stile di vita poco salutare, comprendente una scarsa attività fisica e mentale, un’alimentazione poco equilibrata, l’abuso di alcool o altre sostanze. Il fattore familiarità ha un peso significativo solo in una ridotta percentuale di casi (intorno al 10% per la MA), appartenenti a famiglie portatrici di mutazioni genetiche con numerosi membri affetti dalla malattia, mentre avere un solo parente affetto in una delle generazioni precedenti aumenta solo lievemente il livello di rischio.
Patogenesi
L’ipotesi dominante sulle cause della MA è quella della cosiddetta ‘cascata amiloidea’, cioè di una serie di eventi che hanno come effetto finale la morte dei neuroni cerebrali (neurodegenerazione), innescati dall’accumulo nel cervello della proteina amiloide. Altre forme di demenza neurodegenerativa, per esempio sono sostenute da un meccanismo analogo, ma legato a proteine diverse: l’alfa-sinucleina per la malattia a corpi di Lewy, la proteina Tau o la TDP-43 per le demenze fronto-temporali. In tutte queste patologie la degenerazione neuronale porta ad una riduzione delle dimensioni del cervello in toto o in alcune aree specifiche (es. i lobi frontale e/o temporale), visibile come atrofia all’osservazione macroscopica dell’encefalo.
Per quanto riguarda la demenza vascolare, invece, la disfunzione e la morte neuronali sono legate alla riduzione di apporto di ossigeno e nutrienti e alle reazioni infiammatorie che conseguono al restringimento o all’occlusione (meno frequente alla lacerazione) dei piccoli o grandi vasi che irrorano il tessuto cerebrale. La demenza può comparire dopo un ictus esteso, e in questo caso i sintomi sono direttamente correlati alle funzioni controllate dalle aree cerebrali colpite, oppure svilupparsi nel tempo per l’accumularsi di microlesioni più o meno diffuse.
Sintomi
L’esordio classico della MA consiste nella comparsa insidiosa e progressiva di deficit della capacità di formare nuovi ricordi (memoria di fissazione o anterograda), a fronte di una relativamente conservata capacità di rievocare memorie più o meno remote. Successivamente, nel giro di qualche anno, tendono a comparire difficoltà di orientamento temporale (es. nel riferire la data del giorno) e spaziale, di comprensione e recupero dei vocaboli, di riconoscimento di persone note e di utilizzo degli oggetti, mentre il deficit mnesico diviene progressivamente più severo. In fase avanzata il paziente può non riuscire a distinguere il giorno dalla notte, a riconoscere il domicilio, i propri familiari o anche se stesso allo specchio, ad esprimersi verbalmente in maniera corretta, e a svolgere gesti più o meno complessi. Tutto ciò impatta naturalmente sulla sua capacità di occuparsi della casa, vestirsi e curare l’igiene personale, cucinare, utilizzare il denaro, uscire di casa e spostarsi da solo, assumere correttamente i farmaci, comunicare con gli altri, e così via. Inoltre, anche il movimento e la deambulazione divengono sempre più difficoltosi e incerti. L’aspettativa di vita dalla diagnosi di demenza di Alzheimer è in media di 10 aa circa.
Un altro aspetto clinico molto rilevante, nelle demenze, oltre a quello dei deficit cognitivi e motori, è rappresentato dalle manifestazioni neuropsichiatriche. In fase iniziale sono presenti molto frequentemente depressione, in gran parte reattiva alla diagnosi e alle difficoltà e limitazioni derivanti dalla malattia, e apatia. In alcuni pazienti si sviluppano anche agitazione, irritabilità o aggressività, ed allucinazioni e deliri.
In altre demenze neurodegenerative (ma anche in alcune forme meno tipiche di MA) i sintomi d’esordio non riguardano la sfera della memoria. Nella malattia a corpi di Lewy compaiono in genere deficit visuospaziali ed esecutivi, allucinazioni visive, e disturbi del movimento a tipo malattia di Parkinson: rallentamento nel cammino, rigidità muscolare. Nella demenza frontale comportamentale è soprattutto la personalità a modificarsi, in particolare in senso apatico o francamente disinibito, fatuo, infantile, e poco empatico. Spesso si associa anche un cambiamento delle abitudini alimentari, con tendenza all’iperfagia, al consumo eccessivo di dolci, e talvolta, anche di alcolici. In un’altra forma di demenza frontale e nella demenza del lobo temporale, invece, ad essere coinvolto per primo è il linguaggio. Nella prima forma il paziente diviene progressivamente mutacico, ha un eloquio spontaneo molto ridotto, e compie errori grammaticali e fonemici (scambia lettere o sillabe all’interno delle parole). Nella seconda forma il linguaggio è fluente, ma caratterizzato da anomie, sostituzioni di vocaboli con altri vicini per significato (es. ‘fratello’ per ‘sorella’), e utilizzo di parole generiche (es. coso, quello), e si associano difficoltà di comprensione.
Per quanto concerne la demenza vascolare, ci posso essere deficit di memoria, ma sono soprattutto le funzioni cosiddette ‘esecutive’ ad essere compromesse, con difficoltà di programmazione delle attività, concretezza di pensiero, scarsa capacità di critica e giudizio, difficoltà ad inibire alcuni comportamenti. Particolarmente frequenti e precoci sono l’apatia, la labilità emotiva, le turbe della deambulazione, l’incontinenza. Il decorso clinico è caratteristicamente ‘a gradini’, non progressivo come per le patologie degenerative: a fasi di stabilità dei sintomi seguono bruschi peggioramenti correlati a nuovi eventi di insufficienza circolatoria cerebrale.
Il neurologo rappresenta lo specialista di riferimento per la diagnosi di demenza. Essa si basa innanzitutto sulla presenza dei deficit cognitivi appena descritti, che possono essere oggettivati e quantificati con prove neuropsicologiche specifiche (test di memoria, di linguaggio, visuo-spaziali, di denominazione, ecc. e scale comportamentali). La TAC o Risonanza magnetica dell’encefalo permettono di evidenziare atrofia (inizialmente a livello temporale mesiale/ippocampale, nella MA amnesica, poi diffusa) o esiti vascolari, mentre esami cosiddetti ‘funzionali’, come la PET con glucosio marcato con tracciante radioattivo, rilevano le aree cerebrali meno attive. Negli ultimi anni, poi, per una diagnosi di quasi certezza di MA è possibile eseguire una PET con un tracciante che mette in evidenza direttamente gli accumuli di proteina amiloide a livello dell’encefalo, oppure il dosaggio della proteina nel liquor che circola negli spazi tra le circonvoluzioni cerebrali, che viene prelevato tramite puntura lombare.
Terapia
Al momento vi è disponibilità di trattamento farmacologico per i pazienti affetti da MA, malattia a corpi di Lewy e demenza vascolare, anche se nessuna delle terapie in uso permette di arrestare completamente i processi di danno cerebrale e impedire la progressione dei sintomi. Gli anticolinesterasici (donepezil, rivastigmina, galantamina) e la memantina agiscono potenziando la trasmissione degli impulsi a livello delle sinapsi cerebrali, e sono indicati nelle prime due patologie. Nelle forme vascolari, l’introduzione di vasodilatatori e di farmaci che prevengono le recidive vascolari (es. l’aspirina) migliorano la perfusione cerebrale.
In presenza di disturbi dell’umore e del comportamento, è possibile intervenire con antidepressivi (es. gli inibitori del reuptake della serotonina) e neurolettici, soprattutto quelli di ultima generazione, meno gravati da effetti collaterali cardiovascolari e motori (quetiapina, risperidone, olanzapina).
Infine, anche il controllo dei fattori di rischio vascolare, l’adozione di uno stile di vita attivo e sano (compresa la dieta mediterranea), e gli esercizi di stimolazione cognitiva possono essere di grande beneficio, soprattutto nelle prime fasi di malattia. Promuovere la riduzione del rischio di sviluppare demenza attraverso questi interventi non farmacologici è fondamentale, nell’attesa di trattamenti risolutivi. Sono infatti in corso numerose sperimentazioni, in particolare nel campo della MA in fase di MCI, che, agendo sui processi patogenetici della malattia, puntano a bloccare o prevenire la neurodegenerazione e le sue conseguenze cliniche.
• EPILESSIA
Schede di patologia
Epilessia
La parola epilessia deriva dal verbo greco e′πιλαµβa′νειν (epilambanein) che significa “essere sopraffatti, colti di sorpresa”. L’epilessia è una malattia caratterizzata dalla persistente predisposizione dell’encefalo a generare crisi epilettiche. Quest’ultime sono manifestazioni cliniche molto variegate, che insorgono improvvisamente, hanno breve durata (da pochi secondi a ~2-3 minuti) e sono determinate da impulsi elettrici abnormi di uno o più gruppi di neuroni “ipereccitabili”.
Epidemiologia
L’epilessia è una tra le più frequenti patologie neurologiche, riconosciuta come malattia sociale dall’OMS. Essa può esordire a tutte le età della vita ma i maggiori picchi d’incidenza si hanno nei bambini e negli anziani. L’epilessia interessa circa 1 persona su 100: si stima che in Italia ne siano affette 500.000-600.000 persone.
Patogenesi
Le cause dell’epilessia sono molteplici. Tra le più frequenti si ricordano le anomalie genetiche, i traumi cranici, gli ictus cerebrali, le encefaliti, i tumori cerebrali, le malattie infiammatorie. Ancora oggi, il 30% dell’epilessie ha una causa sconosciuta.
Le crisi epilettiche sono causate da un’alterata funzionalità dei neuroni. Per una delle suddette cause, i neuroni diventano “ipereccitabili” e determinano crisi epilettiche che hanno la tendenza a ripetersi (più o meno frequentemente) nel corso degli anni.
Sintomi
Dal punto di vista clinico, la crisi epilettiche si possono manifestare in maniera estremamente eterogenea in base alla sede di origina dell’eccessiva scarica neuronale. Le crisi epilettiche possono essere generalizzate, se hanno origine da tutti i neuroni dell’encefalo simultaneamente, o focali, se originano in una sede ben circoscritta dell’encefalo.
Le crisi generalizzate si associano quasi sempre a perdita di coscienza. L’esempio paradigmatico è rappresentato dalla crisi tonico-clonico generalizzata (detta anche crisi di “Grande Male”). Questa è caratterizzata da improvvisa perdita di coscienza, emissione di un urlo, caduta a terra, rigidità diffusa (fase tonica) quindi scosse (fase clonica) a tutto il corpo. La persona potrà mordersi la lingua o perdere le urine. Al termine della fase clonica, la persona è generalmente profondamente addormentata (fase post-critica), confusa e, a volte agitata, per un periodo variabile da qualche minuto a qualche ora. Altri tipi di crisi generalizzate, con manifestazioni cliniche meno eclatanti, sono le crisi di assenza (dette anche crisi di “piccolo male”), che si verificano tipicamente nei bambini in età scolare e si caratterizzano per fissità dello sguardo e interruzione dell’attività in corso.
Le crisi focali invece non sempre comportano la perdita di coscienza e si manifestano con sintomi diversi in base all’area cerebrale interessata dalla scarica. Ad esempio, una crisi che inizia nella corteccia occipitale (ove risiede l’area visiva) può comportare la percezione di flash o pallini colorati, mentre una crisi che origina nel lobo frontale (ove è presente l’area motoria) può comportare irrigidimento o scosse dell’emicorpo (volto, arto superiore e/o arto inferiore) controlaterale alla sede di origine della scarica. Le persone affette da crisi focali potranno talora avvertire l’esordio delle crisi e prendere adeguati provvedimenti (sedersi, avvertire gli astanti, etc..). Durante una crisi focale con disturbo di coscienza, il paziente potrebbe sembrare “sveglio” ma non essere in contatto con le altre persone e l’ambiente attorno a sé, non rispondere normalmente agli stimoli esterni e presentare comportamenti ripetitivi (soprattutto alla bocca e alle mani) chiamati “automatismi”.
Avere una crisi epilettica non significa necessariamente essere affetti da epilessia. Molte situazioni possono, infatti, determinare la comparsa di crisi epilettiche isolate, destinate a non ripetersi più spontaneamente (in assenza cioè di quella situazione che le ha generate): l’innalzamento della febbre nei bambini piccoli (fino a 5 anni: “convulsioni febbrili”), l’abuso o l’astinenza da alcool, l’uso di droghe, disturbi metabolici (ad esempio riduzione brusca dei livelli di glicemia, etc..).
La diagnosi di epilessia si basa sulla raccolta completa della storia clinica spesso con l’ausilio dei familiari o di chi ha assistito alle crisi. In seguito, si passa all’esecuzione dell’elettroencefalogramma (EEG) e di indagini quali TAC e/o risonanza magnetica. Altri accertamenti (esami genetici, PET, SPECT) vengono utilizzati in casi selezionati.
Terapia
La terapia dell’epilessia ha l’obiettivo di eliminare o ridurre le crisi e migliorare la qualità di vita delle persone. Il piano di terapia ottimale deve essere stilato a seguito di un’accurata diagnosi e alle caratteristiche della persona affetta (sesso, età, attività lavorativa, etc..).
Il primo approccio alla cura dell’epilessia è basato sull’uso di farmaci antiepilettici. Esistono in commercio oltre 20 farmaci per l’epilessia. Il 70% circa delle persone con epilessia diventa libero da crisi con la terapia farmacologica. Il rimanente 30% non risponde invece alla terapia ed è definito pertanto farmaco-resistente.
Il 15-20% di queste persone farmacoresistenti potrebbe giovarsi di un intervento neurochirurgico, con scomparsa delle crisi nella maggioranza dei casi. La terapia chirurgica consiste nella rimozione della regione del cervello da cui originano e si diffondono le crisi. La selezione dei pazienti candidati all’intervento deve avvenire in modo rigoroso e prevede la costituzione di una equipe multidisciplinare per l’analisi accurata dei dati neurofisiologici (monitoraggio prolungato di video-EEG di superficie, o di profondità con elettrodi intracerebrali, per la registrazione delle crisi), delle neuroimmagini (RMN, PET, ecc), dei test neuropsicologici e delle valutazioni psichiatriche. Alla fine di questo percorso diagnostico si bilancia l’eleggibilità del singolo paziente ad una determinata procedura chirurgica, considerando la causa dell’epilessia, la sede della zona epilettogena (regione del cervello da cui originano e si diffondono le crisi), le sue condizioni cognitive, psicologiche e psichiatriche.
Alcune persone con epilessia farmacoresistente non possono essere sottoposte ad intervento neurochirurgico poiché le crisi originano da molteplici aree nello stesso emisfero o nei due emisferi cerebrali, oppure perché le zone epilettogene coinvolgono aree che non possono essere asportate senza determinare gravi disabilità (es. aree del linguaggio, aree motorie, ecc), o ancora perché le condizioni psicologiche o psichiatriche lo controindicano. In queste situazioni, si può ricorrere a terapie “palliative” (es. stimolazione del nervo vago, dieta chetogenica) che hanno lo scopo di ridurre la frequenza e la gravità delle crisi (senza eliminarle del tutto) ed il numero o la quantità dei farmaci antiepilettici (limitandone quindi gli effetti collaterali), con l’obiettivo di migliorare la qualità della vita.
• ICTUS
Schede di patologia
Ictus
Si definisce ictus una sindrome clinica caratterizzata da comparsa improvvisa di un deficit neurologico focale (più raramente globale), che persiste per più di 24 ore o porta a morte ed è causato da chiusura (ictus ischemico) o rottura (ictus emorragico) di un’arteria cerebrale. Il limite di 24 ore è arbitrario e, secondo alcune definizioni, andrebbe sostituito con un dato di neuroimmagine: cioè se alla TC o alla RM è visibile un’area lesionale si pone diagnosi di ictus anche se i sintomi hanno avuto durata inferiore a 24 ore.
EPIDEMIOLOGIA
Globalmente in tutto il mondo circa 15 milioni di persone sono colpite ogni anno da ictus; tra queste 5 milioni vanno incontro a morte ed altri 5 milioni a disabilità permanente. Quindi, l’ictus rappresenta globalmente la seconda causa di morte e la terza di disabilità. Nei Paesi Europei l’incidenza di ictus varia tra 95 e 290 nuovi casi ogni 100000 abitanti all’anno ed ogni anno vi sono 650000 decessi causati da ictus. Nei Paesi sviluppati l’incidenza dell’ictus si è ridotta del 42% nelle ultime 4 decadi grazie al migliore controllo dei fattori di rischio. L’ictus ischemico rappresenta la maggior parte di tutti gli ictus (65-90%) seguito dalle emorragie intraparenchimali (10-25%) e dalle emorragie subaracnoidee (0,5-5%).
PATOGENESI E FATTORI DI RISCHIO
Alla base dell’ictus ischemico vi è l’occlusione di un’arteria cerebrale, con conseguente riduzione di apporto di sangue e, quindi, di ossigeno e nutrienti al tessuto cerebrale. Questo provoca alterazioni della funzionalità delle cellule cerebrali che conducono rapidamente a necrosi la porzione di tessuto cerebrale più colpita dall’ischemia (il cosiddetto core ischemico), attorno alla quale c’è una porzione di tessuto in sofferenza ischemica ma ancora recuperabile (la cosiddetta penombra ischemica).
L’emorragia cerebrale intraparenchimale viene classicamente distinta in una forma “tipica”, la più frequente, causata da rottura dei piccoli vasi cerebrali profondi conseguente principalmente all’ipertensione arteriosa, ed una forma atipica, spesso secondaria a malformazioni vascolari intracerebrali o a patologie quali l’amiloidosi cerebrale.
L’emorragia subaracnoidea è di norma causata dalla rottura di un aneurisma cerebrale, vale a dire una dilatazione sacciforme della parete malata di un vaso cerebrale.
L’ictus ischemico viene classificato in base alle cause in: cardioembolico, quando il trombo occludente proviene dalle cavità cardiache; aterosclerotico se causato dalla rottura di una placca aterosclerotica delle arterie del collo; lacunare, dovuto ad una patologia dei piccoli vasi cerebrali; da altre cause come dissezione arteriosa, vasculiti, anomalie genetiche ed altre; criptogenetico, ovvero da causa non determinata.
I fattori di rischio per ictus vengono distinti in non modificabili e modificabili.
I fattori di rischio non modificabili comprendono l’età (l’incidenza dell’ictus raddoppia per ogni decade di età dopo i 55 anni), il sesso (l’ictus è più frequente nel sesso maschile fino alla sesta-settima decade di vita, poi aumenta l’incidenza nel sesso femminile anche a causa della maggiore longevità), l’etnia (gli afroamericani e gli ispanici hanno un rischio tra 2 e 4 volte più alto di ictus), i fattori genetici (alcune varianti di specifici loci genici sono state riconosciute come correlate ad aumentato rischio di ictus).
Una storia familiare positiva aumenta globalmente il rischio di ictus di circa il 30%, con maggiore rilevanza nel sesso femminile e nel caso di insorgenza in età inferiore ai 65 anni.
L’identificazione dei fattori di rischio modificabili è di vitale importanza per attuare le misure di prevenzione primaria e secondaria dell’ictus, attraverso interventi farmacologici e modifiche dello stile di vita.
L’ipertensione arteriosa rappresenta il più importante fattore di rischio modificabile per ictus: all’aumento dei valori medi di pressione arteriosa corrisponde un aumento lineare del rischio di ictus. La riduzione della pressione arteriosa sistemica, sia per mezzo di agenti farmacologici che con modifiche allo stile di vita, ha dimostrato di ridurre il rischio di ictus nei pazienti ipertesi.
Il Diabete raddoppia il rischio di eventi cerebrovascolari acuti, con particolare effetto nei pazienti giovani. La correzione farmacologica e con la dieta dei livelli glicemici ha dimostrato di ridurre il rischio di ictus nei pazienti diabetici.
La fibrillazione atriale e la cardiopatia atriale sono importanti fattori di rischio per l’insorgenza di ictus ischemico. Secondo la teoria più classica, la fibrillazione atriale provocherebbe la stasi del sangue nelle camere cardiache, favorendo la formazione di trombi responsabili del cardioembolismo cerebrale e dunque dell’ictus. Tuttavia, più di recente è stato messo in evidenza come non sia propriamente la fibrillazione atriale ma più probabilmente la cardiomiopatia atriale, dunque una condizione strutturale delle pareti cardiache, a provocare il cardioembolismo. Ad ogni modo, in pazienti fibrillanti la prevenzione primaria e secondaria con terapia anticoagulante si è dimostrata efficace nel ridurre il rischio di ictus ischemico.
Il rischio di ictus ischemico ed in particolare id sottotipo aterotrombotico aumenta con livelli elevati di colesterolo e diminuisce all’aumentare del colesterolo HDL. Il trattamento farmacologico ipocolesterolomizzante con statine o con i farmaci anti-PCSK9, riduce il rischio di ictus ischemico senza aumentare il tasso di emorragie cerebrali.
L’inattività fisica correla con diversi effetti negativi sulla salute, incluso un aumentato rischio di ictus. I soggetti che praticano regolarmente l’attività fisica hanno un ridotto rischio di sviluppare patologie cerebrovascolari, probabilmente anche grazie alla riduzione dei valori di pressione arteriosa ed al miglior controllo del diabete.
Per quanto concerne la dieta, è noto che un aumentato importo di sale aumenta il rischio di ictus attraverso l’effetto sui valori di pressione arteriosa. Inoltre la dieta mediterranea, così come una dieta ricca in frutta e vegetali hanno un effetto protettivo.
L’assunzione di alcol ha un effetto diverso a seconda del tipo di ictus. Per quanto concerne la forma ischemica, una moderata assunzione di alcol ha dimostrato un effetto protettivo mentre l’assunzione di elevate quantità di alcol ne aumenta il rischio. Invece, il rischio di ictus emorragico aumenta con l’assunzione di qualsiasi quantità di alcol.
Il fumo di sigarette raddoppia il rischio di insorgenza di ictus con un rapporto lineare tra la quantità di sigarette consumate per giorno e la frequenza di eventi cerebrovascolari acuti. Smettere di fumare riduce il rischio di ictus, eliminando l’effetto negativo del tabacco dopo un periodo di 2-4 anni.
L’abuso di sostanze illegali, quali eroina, cocaina, amfetamine, ecstasy è in relazione con un aumento del rischio di entrambi i tipi di ictus.
È stata studiata la relazione tra i livelli di infiammazione sistemica e alcuni marker ematici come la proteina C reattiva ed il rischio di ictus, tuttavia non è chiaro in che modo l’infiammazione provocherebbe l’insorgenza di eventi cerebrovascolari acuti, se attraverso l’effetto sulla formazione/instabilizzazione di placche aterosclerotiche o tramite un effetto diretto procoagulante. Ad ogni modo, è noto che l’ictus sia ischemico che emorragico può rappresentare la complicanza di alcune patologie infettive quali endocarditi e l’infezione da HIV.
SINTOMI
L’ictus si può manifestare con vari segni e sintomi. Le caratteristiche cliniche che permettono la diagnosi clinica di ictus sono: inizio improvviso, perdita di una funzione focale, sintomi e segni che raggiungono il massimo livello entro pochi secondi o minuti e persistono per più di 24 ore.
L’ictus ischemico provoca una diversa sintomatologia a seconda del territorio vascolare e dunque della porzione di encefalo interessata; i sintomi più frequenti sono deficit motori agli arti controlaterali alla lesione, deviazioni della rima orale, disturbi del linguaggio, disturbi della vista, di sensibilità e dell’equilibrio. L’ictus emorragico oltre ai diversi segni neurologici focali si può manifestare anche con cefalea ad insorgenza acuta, particolarmente intensa nel caso dell’emorragia subaracnoidea ed in quest’ultimo caso associata a rigidità nucale. Frequentemente, i sintomi dell’ictus si associano a caduta a terra del paziente e nei casi più gravi a perdita di coscienza. L’ictus rappresenta anche un’importante causa di declino cognitivo e depressione.
Distinguiamo infine un’altra sindrome definita attacco ischemico transitorio (Transient Ischemic Attack – TIA), caratterizzata dall’insorgenza di sintomi neurologici della durata di meno di 24 ore e senza presenza di infarto cerebrale. I TIA rappresentano quindi la manifestazione di una sofferenza ischemica dell’encefalo senza lo sviluppo di un vero e proprio infarto e comportano un rischio elevato di sviluppare un ictus ischemico nelle settimane e mesi successivi.
TRATTAMENTO
Il trattamento dell’ictus va distinto nelle terapie della fase acuta ed in quelle di prevenzione primaria, cioè nei soggetti che non hanno sofferto di eventi cerebrovascolari e secondaria, mirata a prevenire un nuovo episodio nei pazienti affetti da ictus.
Oltre ai trattamenti specifici per l’ictus ischemico può essere necessario mettere in atto terapie generali di supporto. Queste includono: somministrazione di ossigeno nel paziente ipossico, ventilazione assistita in caso di alterazione di coscienza o compromissione bulbare, correzione di iper/ipotensione, correzione dell’ipoglicemia e trattamento dell’iperpiressia.
È stato dimostrato che il ricovero in fase acuta in reparti altamente specializzati denominati Unità Neurovascolari (Stroke Unit) migliora la prognosi dei pazienti e permette un migliore inquadramento diagnostico e l’attuazione di appropriate misure di prevenzione secondaria, senza prolungare i tempi di ospedalizzazione. Sin dalla fase di ricovero è estremamente importante iniziare precoci trattamenti di riabilitazione neuromotoria e cognitiva.
Le due opzioni di trattamento specifico per l’ictus ischemico acuto sono la trombolisi intravenosa e la trombectomia meccanica. Per entrambi gli approcci, di vitale importanza è il fattore temporale, attualmente inteso non più come un astratto tempo medio di sopravvivenza della penombra ischemica, ma come un tempo individuale definibile grazie all’identificazione di fattori maggiormente legati al paziente come la presenza e la qualità dei circoli collaterali, che consentono un tempo di azione più o meno lungo. In ogni caso, il riconoscimento precoce dei sintomi ed il trasporto immediato in un centro dove si possano attuare le terapie di ricanalizzazione, sono di importanza strategica.
Attualmente in pazienti con ictus ischemico si raccomanda la somministrazione di agenti trombolitici entro 4,5 ore dall’esordio dei sintomi. Il trattamento è altresì raccomandato in pazienti con ictus a insorgenza non nota, qualora la RM in diffusione (Diffusion weigthed – DW) e FLAIR (Fluid Attenuation Inversion Recovery) consentano di datare l’esordio evento almeno entro le 4 ore. È fondamentale iniziare la terapia al più presto, poiché quanto più questa è precoce tanto maggiore sarà il beneficio.
Altra terapia standard dell’ictus ischemico è il trattamento di ricanalizzazione per via endovascolare, vale a dire la rimozione meccanica del trombo occludente dalle arterie cerebrali. La trombectomia meccanica si è dimostrata efficace nel migliorare la prognosi dei pazienti con ictus ischemico da occlusione dei grossi vasi cerebrali entro le 6 ore dall’esordio dei sintomi, in particolare quando associata al trattamento trombolitico endovenoso. Inoltre, studi più recenti hanno evidenziato l’efficacia dei trattamenti meccanici oltre le 6 ore e fino a 24 ore dal teorico esordio dei sintomi (pazienti con ictus al risveglio o esordito in assenza di testimoni, nei quali il teorico esordio dei sintomi viene posto all’ora nella quale il paziente è stato visto/sentito in buona salute per l’ultima volta), in presenza di precisi criteri di selezione definiti con TC di perfusione o con RM DW e PW (Perfusion weighted).
Per quanto concerne la forma emorragica, il trattamento in acuto differisce a seconda della causa dello spandimento emorragico. Anche se con finestre terapeutiche più ampie, anche nell’ictus emorragico è vitale agire il più precocemente possibile.
Per le emorragie dette tipiche, dovute all’ipertensione arteriosa, è necessario attuare trattamenti farmacologici aggressivi mirati a ridurre i valori di pressione arteriosa sistolica al di sotto dei 140 mmHg entro le prime ore dall’esordio dei sintomi. Inoltre possono essere utilizzati farmaci antiedemigeni per ridurre la pressione intracranica e l’effetto compressivo dell’ematoma sul tessuto cerebrale sano. In specifici casi, si pone un’indicazione neurochirurgica per l’evacuazione dell’ematoma cerebrale, al fine di ridurre la pressione intracranica e salvaguardare il tessuto cerebrale.
Nel caso di emorragie conseguenti alla rottura di lesioni vascolari quali aneursmi o malformazioni arterovenose cerebrali, si interviene per via chirurgica o endovascolare, così da ridurre il rischio di risanguinamento, che è particolarmente elevato nelle prime ore dall’esordio dei sintomi, per cui tali interventi dovrebbero essere effettuati non più tardi di 48 ore dall’evento emorragico.
Durante l’ospedalizzazione è necessario uno stretto monitoraggio clinico e strumentale dei pazienti al fine di riconoscere e trattare le possibili complicanze della patologia emorragica, quali vasospasmo, iposodiemia ed idrocefalo, con un rischio particolarmente elevato nelle prime due settimane dall’evento acuto.
I trattamenti di prevenzione consistono in prima istanza nel trattamento dei fattori di rischio modificabili, tramite cambiamenti nello stile di vita ed eventuali terapie farmacologiche.
Nel caso dell’ictus ischemico è necessario anche introdurre terapie antitrombotiche che differiscono a seconda del sottotipo eziopatogenetico: terapia antiaggregante con acido acetilsalicilico o clopidogrel nell’ictus aterotrombotico; terapia anticoagulante orale con i farmaci anti vitamina K o con gli anticoagulanti diretti nell’ictus cardioembolico. Prima di iniziare la suddetta terapia è necessario valutare il rischio emorragico e se questo sia superiore a quello ischemico.
Pazienti con ictus aterotrombotico e placche stenosanti il lume carotideo per il 50% o oltre (stenosi definita in base al rapporto fra diametro del vaso a livello della stenosi e diametro a valle dell’area stenotica) vanno trattati con intervento di endarteriectomia o con apposizione di stent.
Tutti i pazienti che hanno sofferto di un evento cerebrovascolare acuto necessitano di uno stretto follow up, clinico e strumentale, sia neurologico che cadiovascolare ed internistico.
• MALATTIA DI PARKINSON E PARKINSONISMI
Schede di patologia
Malattia di Parkinson e Parkinsonismi
EPIDEMIOLOGIA
La malattia di Parkinson (MP) è il secondo disordine neurodegenerativo, in termini di frequenza, dopo la malattia di Alzheimer. Nei paesi industrializzati ha un’incidenza di circa 12/100000 persone all’anno con una prevalenza, di circa 2 milioni di individui affetti. La malattia è leggermente più frequente nel sesso maschile rispetto al femminile (60% vs 40%).
Colpisce circa l’1% della popolazione con più di 60 anni e raggiunge il 4% tra i soggetti oltre gli 85 anni. Questo suggerisce che un fattore biologico età dipendente, eventualmente in associazione all’esposizione cumulativa ad un fattore ambientale, sia tra gli agenti determinanti.
Nonostante la prevalenza aumenti progressivamente con l’età, non sono rari i casi in cui la malattia si manifesta prima dei 50 anni e anche prima dei 40 anni (Parkinson giovanile).
PATOGENESI
I nuclei della base (nucleo caudato, putamen e globo pallido), un gruppo di strutture cerebrali innervate dal sistema dopaminergico, partecipano alla corretta esecuzione dei movimenti e sono le aree cerebrali più colpite nella MP. Questa si manifesta quando la produzione di dopamina nel cervello cala consistentemente.
I livelli ridotti di dopamina sono dovuti alla degenerazione di neuroni, in un’area chiamata Sostanza Nera (la perdita cellulare è di oltre il 60% all’esordio dei sintomi). Inoltre, nel corso della MP si osserva un progressivo accumulo di una proteina chiamata alfa‐sinucleina neicosiddetti corpi di Lewy. Inizialmente nel bulbo olfattivo, nel midollo allungato e nel tegmento pontino, con i pazienti ancora asintomatici. Col progredire della malattia, i corpi di Lewy si sviluppano nella Sostanza Nera e in altre aree del mesencefalo, nell’ultima fase, nella neocorteccia. Tuttavia, è dibattuto se i corpi di Lewy rappresentino o meno la causa diretta della morte cellulare che caratterizza la MP.
La causa precisa che porta alla MP non è nota ma si ritiene significativa una origine multifattoriale, genetica e ambientale
- Mutazioni genetiche che determinano una MP sono: alfa‐sinucleina (PARK 1), parkina (PARK‐2), PINK1 (PARK‐6), DJ‐1 (PARK‐7), LRRK2 (PARK‐8); altre sono fattori di rischio come la mutazione per glucocerebrosidasi GBA. In generale la MP si esprime qui soprattutto in soggetti giovani. Circa il 20% dei pazienti presenta una storia familiare positiva per la malattia. Si stima che i familiari di persone affette da MP presentino, rispetto alla popolazione generale, un rischio di sviluppare la patologia lievemente superiore.
- Fattori tossici, esposizione lavorativa: il rischio di malattia aumenta con l’esposizione a tossine quali alcuni pesticidi o idrocarburi‐solventi (per esempio la trielina) e in alcune professioni (come quella di saldatore) che espongono i lavoratori a metalli pesanti (ferro, zinco, rame). L’esposizione al fumo di sigaretta riduce probabilmente la comparsa di MP. Il fumo sembra essere cioè un fattore protettivo.
CLINICA
Clinicamente la MP si caratterizza per sintomi motori che vengono comunemente raggruppati nella cosiddetta triade parkinsoniana, tremore a riposo, rigidità plastica e bradi‐ipocinesia. Accanto a queste manifestazioni vanno considerate anche l’instabilità posturale e i disturbi della marcia, che più frequentemente interessano la fase avanzata di malattia. Per porre diagnosi clinica di malattia di Parkinson è necessario presentare bradicinesia e almeno uno degli altri due sintomi che definiscono la triade parkinsoniana. La presentazione tipica dei segni e dei sintomi appena descritti è asimmetrica, generalmente ben obiettivabile all’esame neurologico e ben descritta dal paziente durante la raccolta anamnestica.
Con il termine bradicinesia ci si riferisce alla lentezza del movimento, ovvero all’aumento del tempo di esecuzione del movimento stesso. Agli arti superiori, la bradicinesia si manifesta precocemente con una riduzione della manualità e della gestualità del paziente. I pazienti lamentano difficoltà nell’eseguire compiti semplici, nel portare a termine movimenti piccoli e precisi, come abbottonare vestiti, digitare, legare i lacci delle scarpe. Negli arti inferiori, la bradicinesia appare con un rallentamento della marcia e una riduzione dell’ampiezza del passo, i passi diventano più brevi e piccoli.
Comune è anche la perdita progressiva dei movimenti spontanei (pendolarismo degli arti durante la marcia) e della gestualità e viene a ridursi il linguaggio corporeo che normalmente accompagna il tono emotivo della comunicazione verbale (amimia, inespressività del volto) e la parola subisce cambiamenti significativi, come ipofonia, balbuzie con importanti difficoltà di comunicazione.
La rigidità muscolare può determinare limitazioni funzionali alle articolazioni con dolore associato (tipico è il “blocco” alla spalla).
Il tremore a riposo rappresenta spesso il motivo per cui un paziente richiede un consulto neurologico e nel 70% casi rappresenta effettivamente il sintomo iniziale della MP, sebbene sia assolutamente errato associare questa patologia al solo tremore. Il tremore parkinsoniano è solitamente unilaterale e compare alle estremità degli arti superiori e tende a manifestarsi a riposo o sotto stress emotivo e a scomparire durante l’azione e nel sonno. Per il suo aspetto macroscopico, esso appare simile all’azione del “contar monete”. Il tremore può colpire anche gli arti inferiori e le labbra. Le alterazioni della marcia compaiono piuttosto precocemente nel paziente parkinsoniano, soprattutto nel fenotipo acinetico‐rigido, rispetto al fenotipo prevalentemente tremorigeno.
La marcia è rallentata, a piccoli passi, con particolare difficoltà ad iniziare il movimento (il paziente spesso descrive la sensazione di avere i piedi incollati al suolo, “marcia magnetica”), tendenza alla scomposizione del dietrofront in piccoli passi ed esacerbazione al passaggio in spazi ristretti. In queste situazioni si assiste ad un alto rischio di cadute, rischio aumentato anche a causa della riduzione o completa assenza delle sincinesie degli arti superiori, con conseguente alterata meccanica del passo. Il freezing (o blocco della marcia) è uno dei sintomi più invalidanti della malattia, è tardivo e non è presente nella totalità dei soggetti.
Fase complicata
La fase complicata di malattia si identifica con la severità e con la durata di malattia; è definita dalla comparsa di sintomi legati a modificazioni nella risposta al trattamento con levodopa e dall’associarsi di più sintomi non motori invalidanti. Fluttuazioni motorie di fine dose tipo “wearing‐off” (perdita di efficacia prima della somministrazione successiva) e discinesie fanno parte di queste complicanze. Il peggioramento dei sintomi motori è spesso preceduto o accompagnato da una sintomatologia non‐motoria di agitazione, ansia, disturbi sensoriali e dolore (OFF non motorio). Con il tempo, inoltre, possono comparire intensi OFF notturni, spesso disabilitanti, così come OFF improvvisi e imprevedibili nell’arco della giornata, caratterizzati, a seconda dei casi, da intenso tremore, rigidità e/o distonie dolorose. Le discinesie sono movimenti anomali, involontari.
Possono apparire in vari momenti: al picco dose, all’inizio e fine dose eventualmente accompagnate a distonie. La presenza delle fluttuazioni motorie caratterizza circa il 40% dei pazienti e il maggior rischio di sviluppare fluttuazioni motorie è stato attribuito ad una più giovane età di insorgenza, ad una maggiore severità di malattia e a dosi giornaliere di levodopa più elevate.
Accanto ai sintomi motori, ve ne sono di non motori (NMS, non motor symptoms), i quali sono più insidiosi rispetto ai primi e non meno disabilitanti e invalidanti, gravando negativamente sulla qualità di vita del paziente, soprattutto nella fase avanzata di malattia. I principali sintomi non motori possono essere classificati in disturbi psichiatrici (apatia, depressione, psicosi, sindrome da discontrollo degli impulsi),
cognitivi (demenza), disturbi del sonno (insonnia, parasonnie del sonno REM, in particolare RBD – REM sleep behavior disorders), alterazioni della sensibilità (ipo‐anosmia, ovvero difficoltà a percepire gli odori) e disturbi disautonomici (ipotensione ortostatica, stipsi, scialorrea, incontinenza urinaria, impotenza). La maggior parte di questi insorge in fase avanzata di malattia, tuttavia alcuni di essi compaiono precocemente o possono addirittura precedere di anni, se non decenni, l’esordio della sintomatologia motoria: in particolare, l’anosmia, la stipsi, la depressione e i disturbi comportamentali in sonno REM (RBD) sono reperti frequenti nelle indagini anamnestiche.
TERAPIA
La terapia della MP è sintomatica, e sostanzialmente sostitutiva della carenza di dopamina cerebrale. La levodopa, è il farmaco principale; una volta assorbita nel duodeno attraversa la barriera ematoencefalica e viene trasformata in dopamina nei neuroni della Sostanza Nera. La levodopa viene metabolizzata in periferia dalle dopa decarbossilasi e dalle catecol‐O‐metiltransferasi (COMT). Il blocco di questi enzimi aumenta la concentrazione di levodopa nel sangue e nel cervello e quindi la sua efficacia nel tempo. Le prime vengono inibite dalla carbidopa e dalla benserazide, già associate a levodopa nei farmaci in commercio. Tolcapone, entacapone e opicapone sono invece gli inibitori delle COMT. Un migliore assorbimento intestinale di levodopa si realizza evitando l’assunzione di proteine animali e vegetali a pranzo. I dopamino‐agonisti (DA agonisti) sono farmaci che mimano l’effetto della dopamina (DA) attraverso il legame diretto con il recettore post‐sinaptico della DA. I DA agonisti rappresentano un trattamento efficace per i disturbi motori in tutte le fasi della MP. L’uso dei DA agonisti richiede qualche attenzione: è opportuno informare del rischio di sonnolenza diurna, e in particolare i giovani pazienti, del rischio di compulsione al gioco e allo shopping, all’ipersessualità e all’ aumento dell’appetito (sindrome da discontrollo degli impulsi). Gli inibitori delle monoamino‐ossidasi B (IMAO‐B), selegilina, rasagilina e safinamide, che aggiunge alle prime due un’azione anti glutammatergica, bloccano il catabolismo della dopamina, aumentandone i livelli nello striato. Nella fase avanzata, quando la terapia farmacologica risulta inefficace per garantire al paziente una buona qualità di vita e quando le somministrazioni giornaliere di levodopa superano le 4‐5 dosi e le fasi OFF e le discinesie ON sono sempre più severe, l’infusione continua di un farmaco dopaminergico può rivelarsi un trattamento altamente efficace. Esistono due diversi tipi di terapia infusiva: la somministrazione intestinale di un gel di levodopa/carbidopa e l’infusione continua sottocutanea di apomorfina. La somministrazione intestinale di levodopa tramite PEG/PEJ come quella continua sottocutanea di apomorfina richiede di solito la partecipazione attiva di un “care‐giver”. La stimolazione cerebrale profonda (DBS, Deep Brain Stimulation) rappresenta una efficace e sicura opzione terapeutica per le fasi avanzate della MP. Da ultima risorsa terapeutica come era, si tende oggi a posizionare la DBS nelle fasi intermedie di malattia (early surgery) a fronte di risultati particolarmente positivi. Consiste nell’impianto di due elettrodi alimentati da una batteria, in specifiche strutture del cervello non colpite dalla neuro‐degenerazione, il nucleo subtalamico (STN) e il globo pallido interno (GPi). La scelta del nucleo dipende dalla severità delle fasi OFF e delle discinesie. Gli elettrodi stimolanti, modulano l’attività elettrica, con impulsi variabili per voltaggio (o amperaggio), frequenza e durata (neuromodulazione). Gli avanzamenti tecnologici e l’introduzione di elettrodi direzionali ha migliorato il rapporto tra benefici ed effetti collaterali (finestra terapeutica della DBS). Occorre comunque ricordare ai pazienti che sebbene la DBS sia una tecnica di neurochirurgia funzionale, eventi avversi severi come emorragie cerebrali, per quanto rari sono comunque possibili.
Per la DBS esistono criteri di inclusione ed esclusione, poiché l’efficacia clinica dipende da un accurato processo di selezione del paziente. Il candidato ideale è un paziente di età inferiore a 70 anni con una buona risposta alla levodopa, senza particolari problemi di deambulazione, equilibrio o articolazione della parola, e senza sintomi di decadimento cognitivo.
Una nuova metodica, l’“High Focus Ultrasound‐RMN guidata”, sfrutta gli ultrasuoni per lesioni cerebrali profonde di assoluta precisione. La tecnica ha dimostrato di essere sicura ed efficace sul tremore nelle lesioni talamiche unilaterali; il posizionamento di questa metodica in termini di sicurezza ed efficacia relativamente ai classici target STN e GPi, più utili quindi per la malattia di Parkinson richiede ancora tempo.
I Parkinsonismi Atipici
(PA) sono un gruppo di malattie neurodegenerative sporadiche, caratterizzatedall’associazione di segni parkinsoniani ad altri segni neurologici. Si caratterizzano per un’evoluzione clinica più rapida e per la scarsa o assente risposta alla terapia dopaminergica rispetto alla MP e determinano, di conseguenza una prognosi funzionale più invalidante. Le tre forme principali di PA sono rappresentate dall’atrofia multisistemica (MSA), dalla paralisi sopranucleare progressiva (PSP) e dalla degenerazione corticobasale (CBD). Un neurologo esperto è in grado di individuare alcuni cruciali segni neurologici (in genere: disturbi della motilità oculare ed alterazioni frontali nella PSP, prominenti disturbi disautonomici e/o segni cerebellari e/o piramidali nella MSA, rigidità, mioclono ed aprassia unilaterali nella CBD e l’instabilità
posturale con tendenza alla caduta che si manifesta in tutte e tre le condizioni patologiche) che possono permettere la diagnosi differenziale con la MP già in una fase evolutiva precoce.
• MENINGITI
Schede di patologia
Meningiti
La meningite è un’infiammazione acuta delle leptomeningi (pia madre e aracnoide) e del liquido cefalorachidiano contenuto nello spazio subaracnoideo. Le cause della meningite acuta possono essere infettive, generalmente virus e batteri, e non-infettive, quali disseminazione tumorale, processi autoimmuni, farmaci (meningiti asettiche). Le meningiti batteriche acute sono caratterizzate da infezione purulenta con associata reazione infiammatoria del Sistema Nervoso Centrale (SNC) e rappresentano un’emergenza medica, gravata da un’alta frequenza di complicanze ed esiti neurologici. Per tali motivi è necessaria una diagnosi tempestiva delle meningiti batteriche e l’istituzione di adeguata terapia anti-batterica in tempi molto ristretti.
Negli ultimi anni si è assistito ad un calo nell’incidenza delle meningiti batteriche grazie alle vaccinazioni di fasce di popolazione e di soggetti a rischio, e parallelamente, ad un miglioramento delle tecniche di diagnostica microbiologica e virologica.
Di conseguenza le meningiti virali sono attualmente le forme diagnosticate con maggiore frequenza, anche se, a differenza delle forme batteriche, non esistono terapie antivirali di provata efficacia e mancano studi clinici sulla potenziale efficacia dell’acyclovir nelle forme da herpes.
EPIDEMIOLOGIA
La meningite virale, malattia non notificabile, rappresenta la forma più frequente di meningite con un’incidenza stimata per difetto tra 0,5-17 casi per 100.000 abitanti e una predilezione per soggetti in età infantile. La maggior parte dei casi sono dovuti a Enterovirus non polio, Herpesvirus (HSV-2 e VZV) e Arbovirus (West Nile virus e Toscana virus), questi ultimi con distribuzione geografica confinata a specifiche aree, comprese alcune regioni italiane. Da segnalare l’HIV come causa di meningite nel 25% dei pazienti con infezione primaria, e il virus della parotite nei soggetti non vaccinati.
Le forme batteriche, soggette a denuncia, sono in declino rispetto al passato per effetto dei programmi di immunizzazione su larga scala con vaccini coniugati anti-Streptococco pneumoniae (SP), Neisseria meningitidis (NM), Haemophilus influenzae (HI) tipo b. L’incidenza annuale è di 2,5-6 casi per 100.000 abitanti, con un tasso di mortalità di circa il 14%. Le cause più frequenti nell’adulto sono lo SP, Streptococco di gruppo B, NM, quest’ultima anche sotto forma di epidemie ricorrenti di meningite ogni 8-12 anni, HI, e Listeria monocytogenes. La fonte del contagio con NM sono i portatori sani, e solo in 1 caso su 200 la malattia viene trasmessa da soggetti affetti. Un’importante misura, da osservare sempre, è comunque la sorveglianza dei contatti. Per quanto riguarda lo SP non è necessaria la profilassi dei contatti, in quanto lo stato di portatore è frequente nella popolazione generale e l’infezione si presenta in forma sporadica. La profilassi antibiotica è invece indicata nella meningite da HI nei soggetti con contatto stretto.
PATOGENESI
Le infezioni batteriche dello spazio subaracnoideo possono essere causate da estensione diretta da focolai contigui attraverso le vene della diploe, mediante disseminazione ematogena da focolai distanti, o per cause iatrogene, quali presenza di shunt o derivazioni ventricolari esterne. Nelle forme acquisite in comunità la trasmissione avviene mediante contatto diretto con soggetti portatori o per inalazione mediante la diffusione di goccioline respiratorie o secrezioni nasali. In tali casi, l’evento iniziale è rappresentato dalla colonizzazione batterica a livello nasofaringeo e dalla successiva invasione intravascolare. Lo SP e la NM sono resistenti alla fagocitosi e alla lisi complemento-mediata e dal distretto vascolare possono raggiungere e proliferare nei plessi corioidei e guadagnare l’accesso agli spazi subaracnoidei. In tale sede si assiste a rapida proliferazione batterica con induzione di marcata risposta infiammatoria e rilascio di componenti della parete cellulare batterica, quali lipopolisaccaride acido, acido teoico e peptidoglicani. Queste molecole inducono una marcata reazione infiammatoria meningea con conseguente espressione e rilascio di citochine e chemochine infiammatorie, aumento della permeabilità vascolare e richiamo di cellule infiammatorie circolanti. L’essudato subaracnoideo, composto da leucociti e materiale proteinaceo può compromettere il circolo liquorale e provocare idrocefalo comunicante o ostruttivo. Inoltre, il rilascio di prodotti tossici derivanti dalla degranulazione dei polimorfonucleati neutrofili induce edema citotossico e morte cellulare. Nelle fasi precoci si assiste ad aumento e successivo decremento del flusso cerebrale, fenomeni vasculitici a carico dei vasi subaracnoidei e trombosi dei seni venosi, con conseguente edema vasogenico.
La maggioranza dei virus che causano infezioni cerebrali accede a livello orofaringeo, si moltiplica nei tessuti linfatici e si diffonde nel circolo (fase viremica). L’invasione dello spazio subaracnoideo/tessuto cerebrale avviene attraverso i plessi corioidei, o mediante passaggio diretto o indiretto (linfociti infetti con modalità denominata “cavallo di Troia”) attraverso la barriera emato-encefalica. Raramente l’invasione virale avviene mediante trasporto assonale retrogrado. La presenza dei virus nello spazio subaracnoideo causa una reazione infiammatoria con reclutamento linfocitario e la reazione cellulare è accompagnata da rilascio di citochine infiammatorie e produzione anticorpale da parte delle plasmacellule.
SINTOMI E DIAGNOSI
La meningite batterica può avere un esordio acuto fulminante con progressione nell’arco di ore, specialmente nella popolazione pediatrica, oppure può presentarsi in modo subacuto con peggioramento progressivo nell’arco di qualche giorno, quadro tipicamente osservabile nei soggetti immunodepressi e negli anziani. Febbre, cefalea e rigidità nucale rappresentano la classica triade clinica, alla quale si associa un’alterazione dello stato mentale. La rigidità nucale, caratterizzata da dolore ai movimenti volontari del collo e resistenza passiva alla flessione del collo, ma non generalmente alla rotazione laterale, rappresenta il segno patognomonico dell’irritazione meningea. La rigidità nucale non deve essere confusa con la rigidità cervicale da spondilosi, artrosi e altre patologie osteoarticolari, o con l’ipertono/rigidità cervicale da patologie extrapiramidali, Altri segni classici di irritazione meningea sono il segno di Kernig (evocazione di dolore all’estensione della gamba con paziente supino e coscia flessa sull’addome), e il segno di Brudzinski (flessione alle anche e ginocchia alla flessione passiva del collo. Questi ultimi segni possono essere assenti in varie condizioni, quali grave alterazione dello stato mentale, fasce estreme d’età, immunodepressione, e quindi il loro valore diagnostico è incerto. Un ridotto livello di coscienza, da letargia a stupor o coma, è quasi sempre presente nelle forme batteriche. Sintomi di accompagnamento sono rappresentati da fotofobia, nausea, vomito. Crisi epilettiche sono osservabili nella fase di presentazione clinica o durante l’evoluzione in circa il 25% dei pazienti. Crisi focali suggeriscono la presenza di infarti arteriosi o venosi focali, edema, emorragia, mentre l’occorrenza di crisi generalizzate è secondaria a iperpiressia, iponatriemia, o a cause iatrogene. Altre complicanze sono rappresentate da idrocefalo su base ostruttiva o da alterato riassorbimento liquorale, infarti cerebrali o spinali, neuropatie craniali, perdita dell’udito. L’ipertensione endocranica è una delle complicanze più gravi nelle meningiti batteriche e rappresenta la causa di deterioramento dello stato di coscienza, accompagnato da papilledema, midriasi scarsamente reattiva, paralisi bilaterale del VI nervo cranico, e dal riflesso di Cushing (ipertensione, bradicardia, bradipnea). Un caratteristico rash cutaneo maculo-papulare ad evoluzione petecchiale con interessamento del tronco, arti, congiuntiva e mucose è tipico della meningite meningococcica, forma potenzialmente letale con decesso nell’arco di ore. L’infezione da NM può presentarsi sotto forma di sepsi o come meningite/sepsi e complicarsi con infarto emorragico surrenale bilaterale (sindrome di Waterhouse-Friderichsen).
Nel sospetto di meningite batterica è necessario raccogliere immediatamente campioni di sangue per emocoltura e quindi iniziare tempestivamente una terapia antimicrobica empirica in associazione a desametasone. Per la diagnosi di meningite batterica è assolutamente indispensabile l’esame del liquor cefalorachidiano. Pertanto, una volta escluse controindicazioni e/o la necessità di eseguire esami radiologici, si procede all’esecuzione di puntura lombare con rilevamento della pressione di apertura e la raccolta di campioni da inviare al laboratorio. A differenza dell’emocoltura, che deve essere effettuata su campioni ematici ottenuti prima della somministrazione di antibiotici, I campioni di liquor possono essere ottenuti anche qualche ora dopo l’instaurazione della terapia, senza inficiare i risultati dal punto di vista della tipizzazione citologica dei batteri con colorazione Gram (positività <90%), e la rilevazione degli acidi nucleici batterici mediante “polymerase chain reaction” (PCR). Le alterazioni liquorali nel paziente con meningite batterica sono caratterizzate da aspetto torbido, aumento dei leucociti (da 100 a 50.000/mm3) con prevalenza dei polimorfonucleati neutrofili (>80%), ipoglicorrachia (<40 mg/dL, o rapporto glicorrachia/glicemia <0,4), aumento della concentrazione proteica (>45 mg/dL). Il liquor va inoltre sottoposto ad esame colturale, che risulta positivo in oltre l’80% dei casi, mentre le colture ematiche sono positive nel 60%.
Le meningiti virali sono caratterizzate da cefalea frontale o retro-orbitaria, accentuata dai movimenti oculari, febbre, e irritazione meningea con lieve rigidità nucale. Sintomi di accompagnamento sono rappresentati da mialgie, anoressia, dolori addominali, diarrea, nausea, vomito. Letargia lieve o sonnolenza possono essere presenti, tuttavia raramente si osservano gravi disturbi di coscienza, che quando presenti suggeriscono la presenza di altre cause o di encefalite. Segni neurologici focali o crisi epilettiche sono tipicamente assenti.
L’aspetto del liquor nelle meningiti virali varia da chiaro a opalescente. Inoltre è presente pleiocitosi (50-500 cellule/mm3) a prevalenza linfocitaria e lieve aumento della concentrazione proteica (0.6-09 mg/dL) con glicorrachia normale.
TERAPIA
E’ raccomandabile che il paziente con sospetta meningite batterica inizi immediatamente una terapia empirica combinata con desametasone, seguito a distanza di 15-20 minuti da cefalosporine di terza/quarta generazione e vancomicina. L’ampicillina deve essere aggiunta alla terapia empirica in soggetti con età <3anni e >55 anni, o in pazienti immunodepressi di qualsiasi età per assicurare una copertura contro la Lysteria monocytogenes. Altre combinazioni terapeutiche vanno considerate in casi con neutropenia e in soggetti che hanno subito interventi chirurgici o traumi o nel sospetto di infezione con anaerobi gram-negativi.
Subito dopo la tipizzazione dell’agente patogeno va eseguita terapia specifica.
• NARCOLESSIA
Schede di patologia
Narcolessia
- Narcolessia di tipo 1 (Narcolessia con Cataplessia)
- Narcolessia di tipo 2 (Narcolessia senza Cataplessia)
Epidemiologia
La narcolessia è una rara ipersonnia di origine centrale. Gli studi epidemiologici disponibili, seppure con frequenti limiti imposti dalle metodiche di indagine utilizzata (per lo più questionari e/o intervista clinica, rara disponibilità di conferma polisonnografica, nessuno con disponibilità di dati biologici su sangue o liquor cefalorachidiano), stimano la prevalenza a cavallo di 25-50 su 100000 individui. La malattia può esordire a qualunque età, ma l’esordio più frequente è in età infantile o comunque giovanile, con un picco bifasico in adolescenza e tra i 30-39 anni. La narcolessia presenta un decorso cronico, ma viene per lo più riconosciuta e diagnosticata in genere con grande ritardo stimato in recenti studi mediamente pari a 14-15 anni in casistiche europee e statunitensi. Il grande ritardo diagnostico è considerato possibile causa di una grossolana sottostima della reale prevalenza della malattia, oltre che di un significativo impatto negativo sulla qualità di vita dei pazienti affetti che risultano per lunghi periodi senza adeguata terapia (o addirittura con trattamenti inadeguati causati da misdiagnosi).
Patogenesi
L’eziologia della malattia è considerata complessa e di verosimile origine autoimmune. Dal punto di vista genetico il principale fattore di rischio è la presenza dell’allele HLA-DQB1*0602, ma seppure tale elemento sia estremamente potente solo un portatore su mille di tale gene sviluppa la narcolessia e tale gene è rappresentato in una percentuale variabile tra il 5% ed il 38% della popolazione. Oltre ad altri alleli HLA con impatto sul rischio di sviluppo della malattia, più recenti studi di genetica hanno mostrato un ruolo importante di altre varianti inerenti molecole che interagiscono con il sistema maggiore di istocompatibilità o modulano i processi autoimmuni quali i recettori del T Cell Receptor Alpha e Beta, il Tumor Necrosis Factor Ligand Superfamily Member 4, la pro-catepsina H, il recettore purinergico P2Y 11. In casi straordinari familiari sono state riscontrate rarissime mutazioni considerate causali della malattia.
Fattori ambientali di tipo infettivo sono considerati possibili trigger dello sviluppo della malattia quali la presenza di infezioni delle prime vie aeree, l’influenza da virus H1N1, la vaccinazione con uno specifico vaccino anti influenza H1N1 (Pandemrix), e precedenti infettivi da streptococco pyogenes. L’ipotesi attuale, recentemente avvalorata da studi su processi di immunità cellulo mediata, suggerisce pertanto che un evento trigger infettivo comporti l’attivazione del sistema immunitario determinante poi una aggressione autoimmune sui neuroni ipotalamici producenti orexina/ipocretina. Infatti la malattia sappiamo essere causata dalla scomparsa di tali neuroni nell’ipotalamo dorso laterale, un elemento fisiopatologico che è divenuto di elevata importanza diagnostica essendo l’orexina/ipocretina misurabile direttamente nel liquor cefalorachidiano. Attualmente il deficit liquorale di orexina/ipocretina misurato con tecniche RIA costituisce infatti il marcatore biologico esclusivo della malattia e la narcolessia (di tipo 1) è considerata la sindrome da ipocretino deficienza in vivo.
Sintomi
La narcolessia è caratterizzata da una pentade di sintomi ipnologici a cui possono associarsi una serie di comorbidità mediche. I sintomi inerenti il sonno sono i seguenti: 1) eccessiva sonnolenza diurna con addormentamenti diurni in condizioni inappropriate, per lo più ristoratori ed associati a contenuto onirico, quest’ultima caratteristica intrinsecamente correlata alla rapida emergenza del sonno REM all’addormentamento che risulta essere il marcatore polisonnografico della malattia al test delle latenze multiple dell’addormentamento (MSLT); 2) cataplessia, ovvero episodi di perdita di forza muscolare in veglia con variabile intensità (da una sensazione di debolezza alla paralisi flaccida) e distribuzione (da coinvolgimento focale a generalizzato) che sono evocati da stimoli emotivi, ma in particolare nei pazienti pediatrici o in prossimità dell’esordio dei sintomi possono verificarsi anche senza un trigger (facies cataplettica); 3) paralisi del sonno, ovvero paralisi muscolare propria del sonno REM che si verifica alle transizioni tra veglia e sonno REM (ipnagogiche se all’addormentamento, ipnopompiche se al risveglio); 4) allucinazioni ipnagogiche/ipnopompiche, ovvero commistione di attività onirica all’addormentamento/risveglio determinante fenomeni allucinatori nell’ambiente circostante il paziente; e 5) sonno notturno disturbato dalla presenza di frequenti e protratti risvegli o da concomitanti disturbi motori in sonno quali ad esempio disturbo comportamentale in sonno REM o i movimenti periodici degli arti in sonno. Alla sintomatologia ipnologica si associano spesso comorbidità endocrino-metaboliche (in età adulta obesità, alterazione del controllo glicemico e lipidico; in età infantile obesità ed alterazione dello sviluppo puberale con frequente riscontro di pubertà precoce), cardiovascolari (profilo non-dipper del ritmo della pressione arteriosa), psicologico/psichiatriche (disturbi dell’umore, disturbi d’ansia, disturbi del comportamento alimentare, disturbi dell’ideazione), che in recenti studi sembrano associarsi ad un incremento di mortalità.
Terapia
Seppure siano state segnalati tentativi terapeutici con principi attivi sul sistema immunitario (cortisone, immunoglobuline endovena), attualmente non esiste una cura di provata efficacia per la narcolessia.
Sono disponibili tuttavia numerosi approcci con efficacia sui sintomi della malattia. In primo luogo esistono approcci comportamentali basati sull’igiene del sonno, sui pisolini programmati e sull’attività fisica. Inoltre sono disponibili diversi principi attivi con effetto stimolante, in Italia in particolare sono commercializzati il modafinil (Provigil) ed il pitolisant (Wakix), il primo con effetto dopaminergico ed il secondo con effetto istaminergico. Da segnalare che il pitolisant ha dimostrato efficacia sia sulla eccessiva sonnolenza diurna che sulla cataplessia. Un secondo approccio registrato per la cataplessia è il sodio ossibato (Xyrem), uno sciroppo a base di sale sodico di gamma-idrossibutirrato che si assume di notte; inducendo un sonno ad onde lente risulta efficace su sonnolenza diurna e cataplessia. In Italia il farmaco è registrato per la narcolessia con cataplessia. Infine, seppure off-label, numerosi antidepressivi hanno un potente effetto anticataplettico (e.g. venlafaxina), seppure risentano di un effetto rimbalzo alla sospensione (con possibile induzione di stato cataplettico).
• NEUROPATIE DIABETICHE
Schede di patologia
Neuropatie diabetiche
La Neuropatia Diabetica (ND) è definita dalla presenza di sintomi e segni di disfunzione dei nervi periferici in pazienti affetti da diabete mellito (DM) e in cui sono state escluse altre cause di neuropatia. E’ una delle più frequenti complicanze del DM.
Epidemiologia
Il DM coinvolge circa 435 milioni di persone nel mondo (il 5.9 % della popolazione mondiale) e la sua prevalenza è in continuo aumento. La prevalenza del DM nella popolazione aumenta con l’età. In Italia il 6 % della popolazione dopo i 50 anni è affetto da DM e tale percentuale sale al 20% dopo i 70 anni. Circa metà dei pazienti affetti da DM sviluppa nel corso della malattia una neuropatia che si manifesta con dolore neuropatico nel 15-25% dei casi . La prevalenza della neuropatia nei pazienti diabetici viene stimata tra il 25 % e il 50% in rapporto agli strumenti utilizzati per la diagnosi. Nel “Rochester Neuropathy Study”, che ha incluso tra i criteri diagnostici lo studio elettrofisiologico, è stata individuata una prevalenza della neuropatia del 66% nei pazienti con DM tipo 1 (DM1) e del 59% nei pazienti con DM tipo 2 (DM2). Nei primi la neuropatia si manifesta in almeno il 20% dei casi dopo 20 anni di malattia, mentre nei secondi è presente già al momento della diagnosi di diabete nel 10-15% dei casi e la sua prevalenza sale al 50% dopo 10 anni di malattia.
La ND aumenta il rischio di dolore cronico, cadute, ulcerazioni cutanee, amputazioni e malattie cardiovascolari, impattando pesantemente sulla qualità di vita dei pazienti affetti.
Patogenesi ·
I meccanismi dello sviluppo delle ND non sono ancora completamente chiariti e numerose sono le ipotesi ed i modelli proposti. Verosimilmente, più meccanismi concorrono alla ND.
Secondo la teoria metabolica l’iperglicemia produce un aumento del glucosio intracellulare nel nervo (che come i vasi sanguigni, il cristallino ed i reni non richiede insulina per il trasporto del glucosio) con saturazione della via glicolitica abitualmente utilizzata. Il glucosio in eccesso entra nella via dei polioli e viene convertito in sorbitolo e fruttosio dagli enzimi aldoso-reduttasi e dalla sorbitolo-deidrogenasi. L’accumulo di sorbitolo e di fruttosio conduce ad una riduzione del mioinositolo nel nervo che determina una ridotta attività della pompa Na+/K+-ATPasica di membrana, responsabile di un alterato trasporto assonale e di danno del nervo. Ricerche sperimentali nel topo diabetico hanno dimostrato che l’inibitore dell’aldoso-riduttasi migliora il quadro neuropatico.
La teoria vascolare prevede che la glicosilazione non enzimatica del collagene e di varie proteine delle pareti vasali porti alla formazione dei cosiddetti prodotti finali irreversibili della glicosilazione avanzata (AGE). Questi prodotti si accumulano nelle pareti dei vasi stessi reagendo in modo irreversibile con le proteine plasmatiche producendo fenomeni di aterosclerosi e portando allo sviluppo della microangiopatia diabetica. Inoltre, il legame crociato con componenti insolubili della membrana basale (come la laminina ed i proteoglicani) e della matrice extracellulare, aumenta la permeabilità capillare. Il danno neuronale è pertanto una conseguenza dell’ischemia endonevriale ma anche di un ruolo diretto degli AGE sul metabolismo cellulare, sul trasporto assonale e sulla capacità di riparazione cellulare. Tali effetti sarebbero più pronunciati a livello dei neuroni sensitivi dei gangli dorsali dove la barriera sangue-nervo è incompleta. Gli AGE hanno anche un effetto tossico diretto sulle cellule di Schwann.
La teoria infiammatoria-disimmune si fonda sull’ipotesi che la ND possa essere sostenuta da un meccanismo autoimmune. A sostegno di questa ipotesi vi sono i riscontri bioptici di infiltrati infiammatori localizzati prevalentemente intorno ai vasi peri ed epinevriali del nervo surale prelevato in pazienti diabetici ed i successi terapeutici ottenuti con l’uso delle immunoglobuline endovena.
Infine, sono stati anche ipotizzati una disfunzione mitocondriale e un coinvolgimento dei fattori neurotrofici. In particolare, negli animali diabetici è stata osservata una ridotta produzione ed un rallentato trasporto del Fattore di Crescita Neuronale (NGF)
Sintomi ·
In base al tipo e alla distribuzione del danno nervoso e alle fibre coinvolte, la neuropatia può manifestarsi con diversi quadri clinici diversi e con disturbi che derivano dal prevalente coinvolgimento del sistema nervoso sensitivo, del sistema nervoso vegetativo o del sistema motorio.
Nel 50% dei casi i sintomi sono simmetrici, coinvolgono gli arti distalmente, e sono prevalentemente sensitivi ma possono associarsi deficit di forza e disturbi disautonomici. Questo quadro clinico caratterizza la polineuropatia diabetica simmetrica distale.
Il paziente lamenta dolore che può essere costante o episodico e che descrive come bruciante, come da punture di spilli, a scosse, trafittivo, costrittivo o crampiforme.
Se sono interessate prevalentemente le fibre nervose mieliniche di grosso calibro (responsabili della percezione tattile fine) il paziente lamenta parestesie alle mani e ai piedi (sensazione di intorpidimento e formicolii) e camminando a piedi nudi ha la sensazione che un cuscinetto si interpone tra pianta del piede e pavimento. Percepisce la ridotta sensibilità tattile come la sensazione di toccare gli oggetti attraverso un guanto e questo si traduce in una difficoltà nello svolgimento di attività che coinvolgono i movimenti fini (manipolazione di piccoli oggetti, abbottonare/sbottonare la camicia ecc). Più raramente (nelle forme più gravi) il paziente lamenta una incertezza nella marcia che si rende evidente soprattutto quando viene meno il controllo visivo e quindi al buio o quando chiude gli occhi (atassia sensitiva).
Se la neuropatia è a carico delle fibre sensitive mieliniche di piccolo calibro e amieliniche (neuropatia delle piccole fibre) il paziente lamenta una ridotta capacità di riconoscere stimoli termici e dolorifici e pertanto può tagliarsi o scottarsi senza rendersene conto. Questo quadro di deficit sensitivo può associarsi a sensazioni spontanee simili a punture di “spilli” o aghi, prurito, bruciori, freddo doloroso e fitte dolorose. Tali sintomi spesso peggiorano di notte interferendo con il riposo notturno e talvolta il paziente trova sollievo mettendo i piedi in acqua fredda oppure camminando avanti e indietro per la stanza (sindrome delle gambe senza riposo). Il paziente può lamentare fastidio al contatto con gli indumenti (calze, pigiama o lenzuola (stimolo tattile percepito come doloroso o allodinia meccanica) o riferire eccessivo dolore per uno stimolo nocicettivo di lieve entità (iperalgesia). I disturbi sensitivi possono avere un inizio unilaterale o asimmetrico, ma nelle fasi più avanzate di malattia hanno sempre una distribuzione bilaterale e relativamente simmetrica. Inoltre, i sintomi sensitivi iniziano tipicamente a livello delle dita dei piedi per poi progredire in direzione prossimale, coinvolgendo nel tempo prima gli arti inferiori e poi quelli superiori. Nella maggior parte dei pazienti, l’interessamento delle fibre motorie è modesto e caratterizzato da lieve deficit della forza e del trofismo dei muscoli intrinseci dei piedi.
I sintomi diasutonomici possono essere sfumati e passare inosservati per molto tempo. Il paziente può presentare cute secca agli arti inferiori per un deficit della funzione sudomotoria di cui non si rende conto mentre si lamenta per una eccessiva sudorazione (compensatoria) agli arti superiori e al tronco. La ridotta sudorazione porterà nel tempo a facile affaticabilità ed intolleranza all’esercizio fisico e al caldo. La cute secca distale può essere causa di prurito ed è un fattore predisponente allo sviluppo di ulcere distali (su lesioni da grattamento, ferite e infezioni cutanee). Un aumento della frequenza cardiaca a riposo e soprattutto durante la stazione eretta (tachicardia da intolleranza ortostatica) può indicare un iniziale coinvolgimento del controllo autonomico cardiovascolare e una ridotta funzione del nervo vago, mentre nelle fasi più avanzate di malattia può comparire ipotensione ortostatica. In tal caso il paziente riferisce nel passaggio dalla posizione supina a quella eretta oppure dopo essere stato a lungo in piedi, sensazione di testa leggera e di svenimento (lipotimia) arrivando a collassare nelle fasi più avanzate di malattia. Per l’interessamento delle fibre simpatiche pupillo-dilatatrici possono essere presente miosi e disturbi dell’accomodazione, per cui il paziente lamenta difficoltà a mettere a fuoco soprattutto passando da un ambiente luminoso ad uno più buio. Un alterato controllo autonomico della funzione gastrointestinale è frequente e si manifesta con stipsi e talora con diarrea. Il paziente può presentare anche disturbi urinari (flusso urinario debole, necessità di svuotare la vescica poco dopo aver urinato) e lamentare una disfunzione sessuale come riduzione delle secrezioni e della lubrificazione vaginale nelle donne e difficoltà nell’erezione nel maschio.
La neuropatia autonomica diabetica aumenta il rischio di sviluppo di severi episodi di ipoglicemia perché vengono meno i consueti segnali di allarme (tachicardia, sudorazione) ed è associata ad un maggiore rischio di mortalità per l’insorgenza anche di infarti miocardici silenti (ischemia miocardica senza dolore precordiale) o altre complicanze cardiovascolari.
Oltre ai sintomi descritti che si associano ad un quadro di neuropatia lentamente progressiva, il paziente può presentare sintomi legati ad un interessamento asimmetrico di uno (mononeuropatia) o più nervi (multinevrite) generalmente con un esordio acuto o subacuto. Possono essere colpiti i nervi cranici tra cui più frequentemente il nervo oculomotore; in questo caso il paziente lamenta visione doppia (diplopia) e presenta strabismo e abbassamento della palpebra (ptosi palpebrale).
Forma più rara di neuropatia diabetica è la poliradicolopatia che a sua volta comprende due quadri clinico-patologici, la neuropatia toraco-addominale e la più frequente radicoloplessopatia lombosacrale.
La neuropatia toraco-addominale si presenta in pazienti con età superiore ai 50 anni, è più comune nel DM2 e spesso si associa a perdita significativa del peso corporeo. Si presenta con sensazione di intenso bruciore, senso di costrizione, compressione a cintura, o, infine, di dolore profondo molto più intenso durante la notte, nel territorio di distribuzione delle radici toraciche e/o lombari superiori. E’ inizialmente unilaterale ma frequentemente diventa bilaterale con il progredire della malattia. Nello stesso territorio è apprezzabile un deficit sensitivo.
La radicoloplessopatia lombosacrale, descritta anche come amiotrofia diabetica è presente nello 0.08% dei pazienti diabetici. Si verifica più frequentemente nei pazienti di sesso maschile, con età superiore ai 50 anni e con diabete scarsamente controllato. Una perdita significativa di peso è presente in circa il 50% dei pazienti. I sintomi abitualmente sono unilaterali, ma talora possono estendersi controlateralmente. La sintomatologia è inizialmente caratterizzata da dolore improvviso, severo, unilaterale, localizzato all’inguine e successivamente alla superficie anteriore della coscia. Nei giorni o nelle settimane successive si assiste alla comparsa di ipostenia ed ipotrofia dei muscoli innervati dalle radici lombari più alte (ileopsoas, quadricipite, adduttori) e alla scomparsa del riflesso rotuleo.
Terapia
Non esistono cure specifiche per trattare la ND. Uno stile di vita salutare con una dieta equilibrata e una moderata attività fisica che aiuti a tenere sotto controllo la glicemia e a mantenere il giusto peso corporeo, escludendo altri fattori di rischio (fumo, alcool, eccessivo stress), è fondamentale per rallentarne la progressione e prevenirne le complicanze.
Per la prevenzione del piede diabetico è fondamentale una accurata igiene quotidiana con attenta ispezione della cute e delle unghie, uso di creme idratanti per evitare l’eccessiva secchezza della pelle e uso di calze di cotone e scarpe comode e morbide.
Sono disponibili farmaci sintomatici per trattare il dolore. Questi farmaci appartengono a 2 principali categorie:
-antiepilettici (gabapentin e pregabalin)
-antidepressivi triciclici come l’amitriptilina, o inibitori della ricaptazione della noradrenalina e serotonina (duloxetina, venlafaxina)
-Inoltre sono disponibili analgesici locali da utilizzare quando il dolore è localizzato (Capsaicina, Lidocaina)
Può essere necessario gestire le complicanze relative ai disturbi disautonomici. In particolare:
-Disfunzione Urinaria. Un parziale svuotamento della vescica può portare a ristagno di urine e predisporre ad infezioni ricorrenti. Potrà essere di aiuto, bere adeguatamente (1.5-2 l di acqua al giorno) e svuotare la vescica ad intervalli fissi (ogni 2-3 ore) aiutandosi con manovre facilitanti, ad esempio massaggiando la regione sovrapubica.
-Disfunzione Digestiva. Modifiche della dieta possono essere di aiuto in caso di precoce sazietà (ricorrendo a pasti piccoli e frequenti) o stipsi (aumentando l’idratazione e il contenuto in fibre), diarrea (riducendo il contenuto di grassi e fibre).
-Alterato controllo pressorio. Un calo di pressione arteriosa quando si passa dalla posizione supina a quella eretta o quando si sta in piedi, o dopo i pasti, può essere contrastato con strategie comportamentali come bere 500 ml di acqua 20 min prima di alzarsi dal letto, indossare collant a compressione graduata (140 den) ed eventualmente una panciera, mangiare prevalentemente proteine e verdure limitando l’assunzione dei carboidrati a cena. Ciò contrasterebbe eventuali picchi ipertensivi notturni. A tal fine può giovare anche alzare la testata del letto di 40 cm. Farmaci come la midodrina e il fludrocortisone sono disponibili quando le strategie comportamentali non sono più sufficienti.
-Disfunzione sessuale. Ci sono farmaci che migliorano le prestazioni sessuali ma non sono privi di effetti collaterali, pertanto vengono prescritti dopo accurata valutazione clinica. La secchezza vaginale nelle donne può essere trattata con lubrificanti.
• SCLEROSI MULTIPLA
Schede di patologia
Sclerosi Multipla
La Sclerosi Multipla è una malattia infiammatoria cronica che colpisce il rivestimento mielinico delle fibre nervose a livello del Sistema Nervoso Centrale. La conduzione degli impulsi nervosi e conseguentemente la funzionalità di molteplici aree e sistemi può risultarne compromessa, spiegando la varietà dei sintomi che possono manifestarsi all’esordio o durante il decorso della malattia. Nella maggioranza dei casi (80-85%), la malattia presenta un decorso a “ricadute e remissioni” (forma RR), in cui episodi caratterizzati dalla comparsa di uno o più sintomi di varia natura (visiva, sensitiva, motoria, etc.) si alternano a periodi caratterizzati da assenza di sintomatologia. I sintomi che caratterizzano la ricaduta esordiscono generalmente nell’arco di ore o giorni, durano o si ripetono per almeno 24 ore, e si presentano in assenza di febbre, infezioni o altre malattie concomitanti, andando generalmente incontro a remissione completa o parziale nell’arco di alcune settimane o mesi. Dopo un numero variabile di anni, in assenza di un trattamento specifico in grado di modificare il decorso della malattia, fino all’80% delle forme RR può evolvere in una forma “secondariamente progressiva” (SP) di malattia, in cui, pur potendo ancora presentarsi delle “ricadute” cliniche, il paziente va incontro ad un progressivo peggioramento delle sue funzioni, generalmente corrispondente ad un peggioramento della deambulazione e ad un conseguente aumento della disabilità. In circa il 10-15% dei casi, la malattia presenta invece sin dall’inizio un decorso progressivo (forma “primariamente progressiva”, PP), a cui possono associarsi o meno ricadute.
Caratteristica di questa patologia è la variabilità con cui si manifesta, potendo determinare in tempi successivi (disseminazione temporale) alterazioni delle fibre nervose implicate in diversi sistemi funzionali (disseminazione spaziale), con conseguente ampia variabilità nei sintomi accusati dal paziente e nei segni neurologici osservabili. La diagnosi di Sclerosi Multipla può essere pertanto posta se la disseminazione spaziale e temporale sono presenti e dimostrabili sulla base della storia clinica del paziente, dell’esame obiettivo neurologico e degli esami diagnostici. Tra questi, rivestono particolare importanza la risonanza magnetica (RM) dell’encefalo e del troncoencefalico e del midollo spinale cervicale e dorsale con mezzo di contrasto e l’analisi del liquido cefalo-rachidiano prelevato attraverso la procedura di rachicentesi lombare. Altri esami possono essere di supporto alla diagnosi e utili per escludere altre patologie che potrebbero presentare caratteristiche radiologiche o cliniche simili, come lo studio dei potenziali evocati per la valutazione dell’integrità funzionale delle vie visiva, acustica, somatosensitiva, motoria, in termini di conduzione degli impulsi nervosi. La possibilità di disporre di tali esami permette oggi di formulare la diagnosi di malattia in epoca molto precoce, cioè all’esordio dei primi sintomi, e conseguentemente di instaurare fin da subito una terapia “modificante il decorso della malattia”.
Epidemiologia
La malattia colpisce più di due milioni di persone in tutto il mondo, con una prevalenza 2-3 volte maggiore nel sesso femminile e una maggiore frequenza nella fascia di età compresa fra i 20 e i 40 anni, sebbene siano riportati casi con esordio tardivo o in età pediatrica. La razza caucasica è maggiormente colpita e la prevalenza della malattia nella popolazione aumenta con la latitudine, variando da 100-200 casi ogni 100.000 abitanti nei Paesi del Nord Europa, Nord America e del Sud dell’Australia, a circa 5 casi ogni 100.000 abitanti in Asia ed in Africa nelle regioni tropicali e subtropicali. In Italia, Paese considerato ad alta prevalenza, circa 118.000 persone sono affette da questa patologia ed ogni anno vengono diagnosticati circa 3.400 nuovi casi di Sclerosi Multipla.
Patogenesi
Ad oggi la causa della malattia non è nota, ma si ritiene che, in individui geneticamente predisposti, l’esposizione a determinati fattori ambientali durante l’età dello sviluppo, possa condurre allo sviluppo di una risposta abnorme del sistema immunitario contro proteine specifiche delle cellule nervose. Ciò determina un processo infiammatorio che colpisce la mielina, guaina di rivestimento delle fibre nervose, determinandone la distruzione (demielinizzazione), aspetto predominante nella forma a “ricadute e remissioni” di malattia. Le lesioni conseguenti (“placche di demielinizzazione”) sono più spesso localizzate a livello dei nervi ottici, del tronco encefalico, del cervelletto, del midollo spinale e delle zone “periventricolari” all’interno dell’encefalo. E’ stato tuttavia dimostrato come, accanto al processo di demielinizzazione, si verifichi fin dalle fasi iniziali della malattia una certa sofferenza con perdita delle fibre nervose stesse, che rappresenterebbe l’aspetto predominante se non esclusivo delle forme con decorso progressivo (PP, SP), correlando con l’accumulo di disabilità da parte del paziente. I fattori genetici possono conferire un aumentato rischio di sviluppare la malattia, ma non fanno della Sclerosi Multipla una malattia ereditaria e quindi trasmissibile alla progenie. I fattori ambientali indagati come “responsabili” nell’innesco o nel mantenimento della risposta auto-immunitaria sarebbero molteplici, sebbene non si mai stata trovata una causa definitiva: tra questi, soprattutto l’esposizione a determinati virus, il fumo di sigaretta e bassi valori di vitamina D sembrano poter giocare un ruolo più determinante.
Sintomi
In circa la metà dei casi, i sintomi d’esordio sono determinati dall’alterazione della conduzione nervosa lungo i fasci del midollo spinale, potendo consistere in una perdita di forza più o meno severa o in una riduzione della sensibilità in uno o più arti, nella sensazione di scossa elettrica avvertita alla flessione della testa in avanti, in un’alterazione della sensibilità con formicolii ad un arto, con una distribuzione a fascia a livello del torace o dell’addome, o in tutta la parte inferiore del corpo al di sotto di un determinato livello. Il coinvolgimento della via motoria è responsabile non solo della perdita di forza ad uno o più arti, ma anche della rigidità concomitante, che può presentarsi con spasmi dolorosi e fluttuare o essere costante, limitando ulteriormente il movimento.
Possono anche essere presenti disturbi della funzione vescicale, con difficoltà ad iniziare la minzione o a procrastinarla in presenza dello stimolo o con perdita involontaria di urine, disturbi dell’alvo (stipsi, più raramente incontinenza) e della sfera sessuale (disfunzione erettile, eiaculazione retrograda nell’uomo, anorgasmia nella donna).
Nel 20% circa dei casi, la patologia si manifesta all’esordio con sintomi legati al coinvolgimento della via visiva, più frequentemente della porzione più posteriore del nervo ottico (neurite ottica retrobulbare). Il paziente accusa in questo caso un offuscamento della visione con riduzione dell’acuità visiva in un occhio, difficoltà nella discriminazione dei colori, dolore avvertito dietro o sopra l’orbita che peggiora con i movimenti dell’occhio interessato.
Il coinvolgimento del tronco encefalico può determinare visione doppia, sintomo di esordio fino al 12% dei casi, ma anche riduzione dell’udito, ridotta sensibilità o debolezza dei muscoli in una metà del volto, o improvvisi episodi caratterizzati da intenso dolore trafittivo ad una metà del volto (nevralgia del trigemino).
Nel 10% circa dei casi, i sintomi di esordio della patologia sono espressione della compromissione delle funzioni del cervelletto, comprendendo difficoltà nella coordinazione motoria, perdita di equilibrio nel mantenimento della stazione eretta e nella deambulazione, difficoltà ad articolare la parola, tremore degli arti durante il movimento.
Le alterazioni dell’umore e i disturbi cognitivi raramente rappresentano i sintomi di esordio (nel 5% dei casi), ma possono presentarsi in una maggiore percentuale di pazienti durante il decorso della malattia.
Molto comune tra i pazienti affetti da Sclerosi Multipla è la riferita sensazione di fatica, intesa come una riduzione di energia fisica e mentale con conseguente stanchezza che risulta essere sproporzionata rispetto alle attività svolte e che limita ulteriormente l’adempimento delle attività quotidiane. Tale condizione, analogamente ad altri sintomi della malattia, peggiora notevolmente con l’aumento della temperatura esterna e durante gli stati febbrili, che possono inoltre favorire la comparsa di disturbi transitori, soprattutto visivi o sensitivi, della durata di pochi minuti.
Terapia
Pur non potendo disporre di una terapia risolutiva per la patologia, esistono oggi molti farmaci in grado di “modificare il decorso della malattia” ed approvati per il trattamento della SM forma RR. Questi farmaci non determinano la risoluzione dei sintomi, ma, assunti cronicamente, rallentano il decorso della malattia, riducendo il rischio per il paziente di sviluppare nuove ricadute, nuove lesioni a carico delle aree cerebrali e dei sistemi funzionali e disabilità, evolvendo verso una forma secondariamente progressiva. La terapia più adatta per il paziente affetto verrà scelta dal neurologo tenendo conto di diversi fattori e caratteristiche legate alla malattia del paziente, alla tollerabilità del farmaco, nonché ad eventuali necessità specifiche del paziente (ad esempio, il desiderio di gravidanza).
Nelle forme di malattia lievi-moderate con lento decorso e scarsa attività clinica e radiologica, in pazienti che non presentano elementi prognosticamente negativi, si potrà iniziare il trattamento cronico con una terapia di prima linea, comprendente farmaci che si somministrano per via iniettiva intramuscolare o sottocutanea (interferone beta-1a, interferone beta-1b, glatiramer acetato) o per via orale (dimetilfumarato, teriflunomide).
Ogni farmaco presenta caratteristiche differenti in termini di modalità e tempi di somministrazione, possibili effetti collaterali, compatibilità con desiderio di gravidanza. In alcuni casi i farmaci di prima linea non riescono ad essere efficaci nel contrastare la malattia in maniera adeguata, pertanto sarà necessario adoperare dei farmaci più potenti (farmaci di seconda linea), orali (fingolimod, cladribina) o somministrati con infusione endovenosa in ambiente ospedaliero (natalizumab, alemtuzumab, ocrelizumab), ma con un differente corteo di effetti collaterali.
Alcune forme di malattia possono presentare sin dall’esordio un decorso più aggressivo ed associarsi a fattori demografici e a caratteristiche di malattia predittive di una prognosi più sfavorevole. In questi casi verrà valutata la possibilità di iniziare il trattamento con una terapia d’urto, che sia in grado di bloccare la malattia resettando il sistema immunitario, e successivamente procedere con una terapia di mantenimento. Alcuni farmaci di seconda linea, come la cladribina e l’alemtuzumab, presentano queste caratteristiche, così come il trapianto autologo di cellule staminali del midollo ossero, che può essere proposto in casi accuratamente selezionati.
Per le forme di malattia con decorso progressivo dall’esordio (PP), la prima terapia disponibile (ocrelizumab) è stata approvata nel settembre del 2018. Un nuovo farmaco (siponimod), è stato già approvato in Nord America ed è in corso di valutazione in Europa per il trattamento delle forme secondariamente progressive (SP). In questi casi l’obiettivo della terapia sarà rallentare il decorso della malattia riducendo l’accumulo di disabilità.
Nel caso in cui la malattia sia attiva da un punto di vista clinico, determinando ricadute, la terapia si avvale dell’impiego dei farmaci antinfiammatori corticosteroidei (metilprednisolone) somministrati ad alto dosaggio per alcuni giorni, con lo specifico obiettivo di ridurre l’attività infiammatoria della malattia ed i sintomi che ne derivano. Plasmaferesi ed Immunoglobuline possono essere utilizzati per quei pazienti che non hanno adeguata risposta al metilprednisolone o che presentano controindicazioni assolute all’utilizzo di questo farmaco.
Altri farmaci specifici, sebbene non abbiano effetto sul decorso della malattia, possono essere utilizzati come “sintomatici” per il trattamento dei disturbi accusati dal paziente. La fatica, il sintomo più comunemente riferito dai pazienti, può beneficiare dell’utilizzo di farmaci come l’amantadina, la fampiridina e l’aminopiridina galenica. L’aumento del tono muscolare (spasticità) e gli spasmi muscolari, che colpiscono gli arti interessati dal deficit di forza, possono essere invece trattati con farmaci atti a favorire il rilassamento muscolare, come il baclofene, la tizanidina, il diazepam o le infiltrazioni di tossina botulinica. Nel 2013 è stato inoltre approvato il primo preparato a base di cannabinoidi con indicazione specifica per il trattamento della spasticità (Sativex). Un ruolo fondamentale è svolto dalla fisiochinesiterapia, mirata al potenziamento delle abilità residue, della postura, del mantenimento dell’equilibrio e del cammino, ma anche alla prevenzione delle complicanze legate alla scarsa mobilizzazione ed al mantenimento e miglioramento dell’autonomia del paziente. La riabilitazione cognitiva fa parte della terapia neuroriabiitativa che può essere offerta ai pazienti che presentano certi deficit cognitivi (memoria, attenzione, funzioni esecutive, fra i più frequenti). I disturbi vescicali possono essere alleviati dall’impiego di una terapia farmacologica specifica con anticolinergici o tossina botulinica in caso di minzione urgente o irrefrenabile, cateterismi intermittenti o permanenti per favorire il completo svuotamento della vescica, riabilitazione del pavimento pelvico. Farmaci come il sildenafil e l’alprostadil possono essere utilizzati nel trattamento della disfunzione erettile, così come l’uso di lassativi (ad esempio lattulosio o glicerina) per la stipsi. Anche i sintomi transitori ma a frequente presentazione, prevalentemente sensitivi (dolore, formicolio, crampi) possono essere trattati con farmaci specifici (ad esempio carbamazepina, gabapentin, topiramato), mentre il trattamento dei disturbi dell’umore può essere gestito con farmaci antidepressivi ed ansiolitici.
• SLA
Schede di patologia
SLA
Le malattie del motoneurone sono una famiglia eterogenea di patologie che interessano i neuroni motori: ad esse appartengono la SLA con le sue varianti, la SMA (atrofia muscolare spinale), la malattia di Kennedy, la Sindrome Post-Polio ed numerose altre forme.
La Sclerosi Laterale Amiotrofica (SLA) è la più frequente delle malattie del motoneurone, nota anche come malattia di Charcot (che per primo la descrisse nel 1869, 150 anni fa) o malattia di Lou Gehrig (legenda del baseball americano che si ammalò nel 1939); essa è una malattia neurodegenerativa progressiva, che colpisce i motoneuroni delle corna anteriori del midollo spinale, del tronco encefalico e della corteccia motoria. L’esordio avviene di solito fra i 65 e 75 anni di età ed è spesso subdolo. Il deficit motorio all’esordio può manifestarsi agli arti superiori, agli arti inferiori, a livello dei muscoli a innervazione bulbare o a livello della muscolatura respiratoria.
Essa colpisce circa 3 casi ogni 100.000 abitanti/anno ed ha una prevalenza di circa 10 casi su 100.000 abitanti, inserendola per definizione dell’OMS una malattia rara (pertanto in Italia contiamo circa 5000-6000 malati). Ancora oggi, seppur molto meno, il sesso maschile ha un’incidenza maggiore di quello femminile. Molti fattori di rischio ambientali sono stati studiati in associazione alla malattia: il fumo di sigaretta sembra essere, insieme all’attività fisica intensa (sportiva o lavorativa) ed ai traumi generali, il fattore di rischio statisticamente più rilevante.
Le cause della malattia sono ancora sconosciute. Negli ultimi anni è stato riconosciuto un ruolo sempre più importante alla genetica, come possibile fattore causale o predisponente della malattia. La SLA è infatti per lo più considerata una patologia sporadica (85-90% dei casi), sebbene esistano forme familiari (10-15% dei casi). Si conoscono ad oggi quattro geni maggiori coinvolti (SOD1, TARDBP, FUS, c9orf72) e numerosi geni minori (ALS2, SETX, VAPB, FIG4, ERBB4, MATR3, ANG, OPTN, VCP, UBQLN2, CHMP2B, PFN1, hNRPA1 A2/B1, TUBA4A, NEK1, SQSTM1). Le mutazioni identificate sono responsabili di circa il 60% dei casi familiari e circa il 12% dei casi sporadici.
Recentemente, grazie allo studio dei dati derivanti dai registri di popolazione, sono stati definiti diversi fenotipi di malattia, dalla SLA classica, alla forma bulbare pura, alla sclerosi laterale primaria (PLS o malattia del I motoneurone), alla atrofia muscolare progressiva (PMA o malattia del II motoneurone), alla forma denominata ‘flail arm syndrome’ (sindrome dell’uomo nel barile) alla ‘flail leg syndrome’ (sindorme delle gambe flccide), alla SLA monomelica.
Sempre nelle ultime due decadi è emerso il ruolo dei deficit cognitivi, che sono presenti, in ogni loro aspetto, fino al 50% dei casi; dalla demenza fronto-temporale classica (10-15% dei casi) alle sindromi disesecutive e alle forme comportamentali, ponendo l’attenzione su un concetto nuovo, quello dello ‘spettro di malattia’, in un continuum tra forme motorie pure a forme cognitive pure.
L’aspettativa di vita dall’esordio dei sintomi è mediamente di 3-5 anni, ma il decorso della malattia varia da paziente a paziente.
Data la notevole eterogeneità di questa patologia, la diagnosi risulta complessa ed è tuttora da considerarsi clinica, sebbene ci siano alcuni importanti contributi dalla neurofisiologia, dalle tecniche di neuroimaging (tecniche avanzate di risonanza magnetica nucleare o delle utilizzo della PET cerebrale).
Inoltre non esistono terapie farmacologiche in grado di arrestare il suo decorso. Ad oggi, l’unico farmaco approvato per la SLA, con efficacia dimostrata in diversi studi controllati, è il riluzolo, capace di rallentare il decorso della malattia di alcuni mesi riducendo l’azione eccitotossica del glutammato. In Italia è autorizzato (non approvato) un altro farmaco, l’edaravone endovenoso, che ha mostrato alcuni risultati sul rallentamento di perdita di funzione.
Tuttavia, grazie al supporto di ausili tecnologici, alla maggiore consapevolezza dei bisogni dei pazienti e al sorgere di centri clinici specializzati, nel corso degli anni la qualità di vita dei malati è decisamente migliorata. Importante ruolo in questo lo svolgono le tecniche di nutrizione enterale supplementare via Peg o RIG, e le tecniche di ventilazione meccanica non-invasiva o invasiva.
I fattori prognostici positivi sono l’età giovanile, il ritardo diagnostico (più è rapida la diagnosi, peggiore è la prognosi), alcune mutazioni genetiche, l’essere seguiti da centri specializzati (fattore che migliora la prognosi), la presenza di deficit cognitivi ( fattore che riduce l’aspettativa di vita).
Il modello utilizzato dai suddetti ‘Centri SLA’ è quello delle cure simultanee, che si basano sulla interdisciplinarietà della presa in carico dei pazienti, in maniera che tutte le figure professionali coinvolte nel ‘team’ lavorino di concerto e pongano al centro dell’intervento terapeutico il malato e la sua famiglia.
La SLA è un ‘paradigma’ nell’ambito delle malattie neurodegenerative, poiché è a causa sconosciuta, ha carattere rapidamente progressivo, causa una graduale perdita dell’autonomia e riduce l’aspettativa di vita: pertanto per l’individuo affetto dalla patologia si pongono numerosi dilemmi etici, in particolare riguardanti le misure di sostentamento vitale.
In questo senso, i malati di SLA, le loro famiglie e i neurologi curanti, si trovano di fronte ad affrontare il tema delle ‘scelte’: da sempre il paziente affetto da questa malattia viene posto di fronte all’espressione delle ‘volontà’ o DAT (disposizioni anticipate di trattamento; oggi, dopo l’entrata in vigore della legge 219, gli strumenti di legislativi sono nettamente migliorati.
Nonostante la mancanza di nozioni sulle esatte cause di malattia, la ricerca ha attualmente grande impulso: gli studi sulla genetica stanno rivelando un numero sempre più importante di geni associati alla SLA; grande impulso hanno recentemente i progetti volti all’identificazione dei marcatori diagnostici e prognostici, dagli ‘staging systems’ ai marcatori sierici e liquorali come i neurofilamenti, o le tecniche di neuroimaging.
Accanto a tali filoni di ricerca, sono attivi numerosi ‘trials’ terapeutici (sperimentazioni), con farmaci, tecniche innovative e d approcci di terapie cellulari: in questo campo molti membri del Gruppo di Studio della SIN sono promotori o partecipanti attivi.
• TUMORI CEREBRALI
Schede di patologia
Tumori cerebrali
Le neoplasie cerebrali sono forme tumorali che colpiscono il sistema nervoso I tumori cerebrali vengono classificati in primitivi e secondari. I primitivi originano all’interno del sistema nervoso da cellule di origine nervosa, mentre i secondari sono frutto di una diffusione metastatica a livello cerebrale di neoplasie sistemiche. I tumori cerebrali secondari (metastasi) rappresentano da soli circa la metà di tutte le neoplasie cerebrali. Le neoplasie sistemiche che più frequentemente danno metastasi encefaliche sono: polmonari, mammarie ed il melanoma. Gliomi, metastasi, meningiomi, adenomi pituitari e neuromi del nervo acustico rappresentano il 95% di tutti i tumori cerebrali dell’adulto. Nei bambini, i tumori cerebrali rappresentano il tumore solido più diffuso, secondo solo alla leucemia. Il tasso di incidenza delle neoplasie primarie del SNC è di 3,6 casi per 100.000 bambini ogni anno.
L’incidenza annua dei tumori cerebrali primitivi è compresa tra i 7-19,1 casi per 100.000 abitanti. I tumori metastatici sono più comuni, si stima che negli USA siano diagnosticati ogni anno circa 200.000 nuovi casi. La diagnosi di tumore cerebrale è lievemente più frequente nell’uomo (rapporto tra maschio e femmina di 1,5:1), tuttavia meningiomi e adenomi pituitari sono leggermente più comuni nelle donne che negli uomini. Per quanto riguarda la localizzazione dei tumori cerebrali I tumori nella fossa cranica posteriore predominano nei bambini, mentre l’incidenza di tumori localizzati al di sopra del tentorio (sovratentoriali) aumenta dall’adolescenza all’età adulta. I gliomi a basso grado di malignità, come l’astrocitoma, sono più comuni nei giovani rispetto all’ anziano. I gliomi ad alto grado di malignità, come l’astrocitoma anaplastico ed il glioblastoma multiforme, tendono ad essere più comuni nel quarto e quinto decennio di vita.
L’esame istologico include oggi ( diagnosi integrata) la definizione del tipo di tumore, il grado ed indagini molecolari che aiutano a predire l’aggressività del tumore, la sua prognosi nonché risposta ai trattamenti
Per i tumori gliali, il grado è determinato da alcune caratteristiche, che emergono all’esame microscopico. Nei tumori gliali la gradazione del tumore parte dal Grado I ( es astrocitomi pilocitici) in cui sono raggruppati tumori caratterizzati da una crescita lenta con modesti aspetti infiltrativi. All’opposto il grado IV esprime il massimo grado di malignità ( glioblastoma) in cui il tumore presenta elementi cellulari altamente proliferanti, proliferazione di vasi associati ad aree di necrosi con un marcato aspetto infiltrativo.
Per quanto riguarda lo studio dei fattori di rischio ad oggi non sono noti fattori inequivocabilmente associati allo sviluppo delle neoplasie cerebrali. Unica eccezione, le radiazioni ionizzanti associate allo sviluppo di meningiomi. Alcune malattie ereditarie, come la: neurofibromatosi, la sclerosi tuberosa, nevoid basal cell carcinoma syndrome, la sindrome di , Turcot, la sindrome di Li-Fraumeni ( mutazione TP53) e la malattia von Hippel-Lindau disease. e; sono associate ad un maggior rischio per sviluppo di neoplasie del SNC.
Manifestazioni cliniche
Le manifestazioni di un tumore cerebrale dipendono soprattutto dalla sua localizzazione e dalle sue dimensioni . Le manifestazioni cliniche in pazienti con neoplasia cerebrale tendono ad essere simili per i tumori cerebrali primitivi e per le metastasi. I sintomi e segni dipendono all’azione svolta dal tumore nel suo accrescimento, che può portare a: compressione , infiltrazione del tessuto sano, dislocazioni delle componenti cerebrali, aumento della pressione intracranica.
L’insorgenza dei sintomi di solito è insidiosa. La sintomatologia può non essere specifica e comprendere: cefalea, nausea, vomito, alterazioni dello psichismo, disturbi dell’equilibrio ( atassia), disturbi del linguaggio ( afasia), deficit della forza ( ipostenia) degli arti o disturbi delle sensibilità ( ipoestesia). Talora la neoplasia rappresenta un reperto occasionale scoperto durante l’esecuzione di esami radiologici per altre motivazioni.
Le crisi epilettiche, focali o generalizzate, possono essere la prima espressione di un tumore cerebrale. Le crisi epilettiche possono essere presenti da mesi o da a anni prima che venga diagnosticato il tumore cerebrale. Nei tumori gliali ( gliomi) le crisi epilettiche sono il primo sintomo nel 60 % dei pazienti, sono più frequenti nei tumori meno aggressivi ( gliomi di basso grado) e nei giovani.
Per tutti i pazienti che sviluppino una prima crisi epilettica in età matura deve essere presa in considerazione la possibilità di un neoplasia cerebrale.
Un esordio che suggerisca un evento vascolare acuto ( ictus) rappresenta una evenienza meno frequente.
Diagnosi
In tutti i pazienti che abbiano sviluppato una sintomatologia suggestiva per neoplasia cerebrale è necessario prendere in considerazione l’esecuzione di accertamenti neuro-radiologici,La risonanza magnetica con contrasto (RM) è la modalità diagnostica di prima scelta. Qualora non sia possibile eseguire una risonanza (ad es. in pazienti con impianti metallici, dispositivi incorporati o claustrofobia), viene utilizzata, pur non avendo la stessa risoluzione della RM, la tomografia computerizzata (TC). Abbiamo poi a disposizione altre tecniche radiologiche dette avanzate come le tecniche di perfusione e spettroscopia della risonanza magnetica o PET ( positron emission tomography) che possono rappresentare una utile integrazione degli strumenti diagnostici a nostra disposizione. Benché la TC e la RMN siano esami fondamentali non solo per la diagnosi, ma anche per programmare l’intervento chirurgico o la radioterapia, la certezza della diagnosi si ha solo con l’esame istologico sia esso derivante da una biopsia sia da un intervento di aspotazione della neoplasia
Il trattamento
La presa in carico dei pazienti affetti da neoplasia cerebrale dovrebbe essere eseguita da un gruppo multidisciplinare che comprenda: un neurochirurgo, un neuro-oncologo, un neuro-radiologo ed un radioterapista esperto di trattamenti sul SNC. L’approccio terapeutico al tumore varia considerevolmente in funzione dell’età del paziente, della sede della neoplasia. dall’aspetto della neoplasia stessa che può suggerire una maggiore o minore malignità e dalla presenza di comorbidità.
Le opzioni di trattamento chirurgico possono includere la rimozione del tumore più o meno completa o la biopsia..
Qualora un paziente con neoplasia cerebrale lamenti cefalea persistente, eventualmente accompagnata da vomito, od un peggioramento dei sintomi è probabile che alla base del deterioramento clinico vi sia un aumento dell’edema che accompagna lo sviluppo della neoplasia. I corticosteroidi possono ridurre drammaticamente questi i segni e sintomi Il desametazone è lo steroide più utilizzato.
La radioterapia (RT) ha un ruolo privilegiato nel trattamento dei tumori cerebrali primitivi e delle metastasi. I progressi nell’imaging e delle tecniche di radioterapia hanno permesso una più precisa localizzazione tumorale, permettendo una riduzione del volume di tessuto cerebrale sano irradiato, con una riduzione del rischio di potenziali tossicità a lungo termine della radioterapia pur mantenendo la sua efficacia.
La chemioterapia può risultare utile in alcuni tipi di tumore cerebrale. Un ostacolo nel trattamento chemioterapico dei tumori cerebrali è rappresentato dalla presenza della barriera emato-encefalica . Questa è una sorta di filtro naturale che limita il passaggio delle sostanze e quindi anche dei farmaci, dal sangue ai tessuti cerebrali. Tra i farmaci in grado di attraversare la BEE quelli che vengono più frequentemente utilizzati sono gli alchilanti come la Temozolomide e le nitrosouree.
Per i pazienti affetti da glioblastoma ( WHO IV) e più in generale glioma di alto grado (WHO III), lo standard di cura è rappresentato dalla combinazione di radioterapia con Temozolomide somministrata a bassi dosaggio, quotidianamente e per tutta la durata della radioterapia ( chemioterapia concomitante) . Alla conclusione della radioterapia, dopo un mese di pausa terapeutica il Paziente inizia una assunzione mensile di temozolomide ( chemioterapia adiuvante) per un periodo che in genere varia da 6 a 12 mesi.
La combinazione di 3 farmaci, Lomustina, procarbazina, e vincristina (PCV), utilizzati in modo sequenziale dopo la radioterapia ha contribuito ad allungare in modo significativo la sopravvivenza di pazienti affetti da oligodendroglioma di grado III con una co-delezione 1p19q. La stessa combinazione si è dimostrata efficace nel prolungare la sopravvivenza di pazienti affetti da gliomi di basso grado quando somministrata dopo la radioterapia. Sono inoltre in corso studi clinici che vedono l’uso della chemioterapia per ritardare la radioterapia nei pazienti affetti da glioma di basso grado.
I pazienti mentre ricevono un trattamento attivo abitualmente, sono monitorati con una risonanza magnetica cerebrale ogni 2 o 3 mesi. L’intervallo del tempo tra le RM aumenta a seconda del grado del tumore.
Per supporto ed assistenza:
Segreteria SIN – SienaCongress
Via del Rastrello, 7 – 53100 Siena
Tel. 0577 286003
info@neuro.it
RESTIAMO IN CONTATTO
![]()
![]()
![]()
![]()
© 2025 Società Italiana di Neurologia
Tutti i diritti riservati.
Cookie Policy
Realizzazione: STUDIOVISIO